|
Una calibración implica obtener los incrementos
de las propiedades termodinámicas de la reacción entre las fases puras que
permiten reproducir la dependencia P-T-X de la constante de equilibrio observada
en sistemas experimentales y/o naturales. Por lo general, los calibrados
presentan restricciones de aplicabilidad resultantes de los rangos P-T-X bajo
los que se han analizado experimental o empíricamente los sistemas. Un elemental
sentido de precaución sugiere calificar con reservas los resultados obtenidos a
partir de extrapolaciones de los calibrados, ya que la linearidad de la ecuación
(49) es una simplificación aplicable a rangos P-T-X determinados.
Para ejemplificar los métodos de calibrado y
evaluar la significación de las constantes que aparecen en las ecuaciones
propuestas en la literatura y sus limitaciones de aplicabilidad, seguiremos
considerando los dos ejemplos anteriormente utilizados, el equilibrio de
intercambio FeMg-1 biotita-granate (termómetro GARB) y el equilibrio
de transferencia neta entre los componentes Al2SiO5,
anortita, grosularia y SiO2 en las fase aluminosilicato, plagioclasa,
granate y cuarzo (barómetro GASP).
El termómetro basado en la reacción de
intercambio Fe-Mg entre granate y biotita, puede expresarse mediante la reacción
entre los componentes de fase almandino (alm), piropo (prp), annita (ann) y
flogopita (phl) (ec. 37; ver Tabla 1 para referencias). Ambas soluciones sólidas
pueden describirse mediante modelos de mezcla generales, de tipo iónico
multiposicional, para los que la actividad se define como (Tema 3 del programa
de Petrogénesis Metamórfica I. Aproximación Termodinámica):
ak =
Õ[(Xi·gi)]ni
(52)
donde el producto
aplica a todas las posiciones estructurales donde ocurre la mezcla, Xi
y gi son las
fracciones molares y coeficientes de actividad posicionales,
respectivamente, de los componentes elementales en las distintas posiciones
estructurales, y ni es la multiplicidad de estas posiciones. Si
aplicamos estos modelos de solución generales a granate y biotita, las
actividades de almandino y annita, por ejemplo, son:
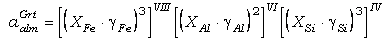 (53)
(53)
 (54)
(54)
donde la mezcla se
produce en todas las posiciones estructurales, indicadadas por la coordinación
en números romanos,
excepto en las ocupadas por el oxígeno. Si se sustituyen las expresiones (53) y
(54), y las expresiones equivalentes para las actividades de piropo y flogopita,
en la ecuación (23) para el equilibrio de intercambio biotita-granate (ec. 37),
reajustando y simplificando, obtenemos:
 (55)
(55)
En este punto, merecen
destacarse dos aspectos de las reacciones de intercambio.
En primer lugar,
las actividades posicionales de los componentes elementales que no están
implicados directamente en la reacción de intercambio se cancelan, por lo que no
aparecen en la expresión de la constante de equilibrio. Esta simplificación es
una ventaja en el tratamiento de este tipo de equilibrios, que no ocurre en el
caso de las reacciones de transferencia neta.
En segundo lugar,
el balance estequiométrico de la reacción de intercambio condiciona el hecho de
que todos los términos de Keq están elevados a una misma potencia,
que es igual a los moles de iones intercambiados (en este caso 3). Por tanto,
puede recurrirse a una simplificación ulterior si consideramos un mol de ión
intercambiado, esto es reescribiendo la reacción de intercambio (37):
1/3 Fe3Al2Si3O12
(alm) + 1/3 KMg3AlSi3O10(OH)2
(phl)
= 1/3 Mg3Al2Si3O12
(prp) + 1/3 KFe3AlSi3O10(OH)2
(ann) (37b)
de manera que los
exponentes, i.e., las multipliciadades de las posiciones estructurales (en este
caso 3) y los coeficientes estequiométricos de los componentes moleculares (en
este caso 1/3), se cancelan:
 (56)
(56)
y los términos de la
constante de equilibrio aparecen con exponente 1. Esta cancelación de los
exponentes en los términos de Keq afecta a cualquier reacción de
intercambio, incluso en el caso de que las multiplicidades de las posiciones
estructurales y los coeficientes estequiométricos de los componentes moleculares
sean distintos en las dos fases, como por ejemplo en el caso del intercambio Fe-Mg
entre granate y clinopiroxeno (diópsido-hedenbergita; ver Tabla 1):
1/3 Fe3Al2Si3O12
(alm) + CaMgSi2O6 (di) =
= 1/3 Mg3Al2Si3O12
(prp) + CaFeSi2O6 (hd) (57)
La simplificación de Keq realizada
anteriormente es conveniente desde el punto de vista del tratamiento analítico
de los equilibrios,
ya que permite identificar la parte ideal de la constante de equilibrio con el
coeficiente de distribución o reparto KD de dos
componentes entre dos fases. Para el intercambio Fe-Mg entre biotita y granate,
y teniendo en cuenta que Xk = nk/Snk,
la ecuación (56) queda:
 (58)
(58)
donde Kg
es el producto de los coeficientes de actividad posicionales.
Nótese que si la reacción de intercambio se
define como en (37), con tres moles de ión intercambiado, entonces la relación
entre la constante de equilibrio y el coeficiente de reparto es Keq=KD3.
Las propiedades termodinámicas de granate y biotita son tales que el granate
fracciona preferentemente el Fe respecto de la biotita. En consecuencia, los
valores de KD serán siempre menores de 1 si la reacción de
intercambio se escribe como en (37), ya que (Mg/Fe)Grt<(Mg/Fe)Bt
(o XMgGrt<XMgBt). Sin embargo, debe
notarse que si la reacción de intercambio se escribe en sentido contrario que en
la ecuación (37), los valores de KD serán mayores de 1, i.e., iguales
al inverso del KD para la reacción escrita como en (37).
El incremento de energía libre de la reacción entre los componentes puros (DGro(P,T))
presentará en cualquier caso el mismo valor absoluto, pero el signo variará en
función de cómo se escriba la reacción.
En la expresión (58), el coeficiente de reparto
KD se ha expresado de formas distintas, llegándose a la formulación
más simple como una razón de las razones Mg/Fe de cada fase. No obstante, esta
formulación no es siempre posible. En el caso anterior puede llegarse a ella
porque los elementos Fe y Mg se encuentra en una única posición estructural en
las fases biotita (posición octaédrica) y granate (posición cúbica
distorsionada). Sin embargo, cuando esto no es así la identificación de la parte
ideal de Keq con KD no es posible. Este punto puede
clarificarse considerando la reacción de intercambio Fe-Mg entre granate y
clionopiroxeno (57). La fórmula unidad ideal del clinopiroxeno es Ca[M2](Fe,Mg)[M1]Si2O6,
de manera que Ca y (Fe,Mg) están restrigidos a dos posiciones estructurales no
equivalentes (M1 y M2). Siguiendo la argumentación anterior tenemos:
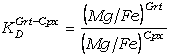 (59)
(59)
Sin embargo, los
piroxenos naturales son, generalmente, deficitarios en Ca, y la posición M2 está
parcialmente ocupada por (Fe,Mg), por lo que Fe[M1]
¹ Fetotal
y Mg[M1]
¹ Mgtotal,
y por lo tanto Keq ¹
KD:
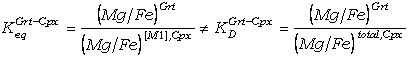 (60)
(60)
Es interesante considerar aquí la fraccionación
entre dos fases de un componente diluido (e.g., traza) que sigue la ley de
Henry. Este comentario se introduce por la posible utilidad en termobarometría
de equilibrios entre soluciones diluidas, que a priori presentan menos problemas
teóricos al poder ser considerados los coeficientes de actividad de los
componentes diluidos como constantes (ley de Henry). La denominada ley de
distribución de Nernst para la fraccionación de un componente diluido puede
ser derivada como un caso especial de la expresión de KD. Así, en el
caso de soluciones binarias que intercambian dos componentes 1 y 2, el
coeficiente de reparto puede escribirse como:
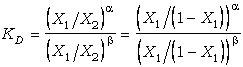 (61)
(61)
Si uno de los
componentes intercambiados (e.g., el componente 1) está diluido en las dos
fases, entonces (1¾X1)
» 1 y KD
» X1a/X1b.
Por otro lado, si se obedece la ley de Henry,
g1=constante, y
g2=1, por lo
que, para condiciones P-T fijas:
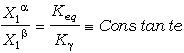 (61)
(61)
que es la ley de
distribución de Nernst. Dado que Kg
no varía con la composición, la partición de elementos en traza ofrece claras
ventajas respecto de la partición de elementos mayores, cuyo comportamiento es
generalmente no ideal, por lo que existen dificultades para definir las
dependencias P-T-X de sus coeficientes de actividad. Sin embargo, hay que notar
que en el caso de soluciones multicomponentes,
g1 puede no ser
constante bajo concentraciones diluidas si varían las proporciones de otros
componentes. El efecto de las variaciones composicionales del solvente sobre el
coeficiente de actividad, y sobre la fracionación, de un componente traza será
pronunciada si los componentes del solvente se comportan de forma no ideal
(i.e., no siguen la ley de Raoult). Por tanto, la ley de distribución de Nerst,
y su posible aplicación en termobarometría, es sólamente válida para soluciones
binarias, o para soluciones multicomponentes que se comporten como binarias,
esto es, aquellas en las que las porporciones de los componentes del solvente se
mantengan fijas.
La fraccionación elemental entre dos fases
puede evaluarse gráficamente considerando la expresión del coeficiente de
reparto como la razón de razones elementales en las dos fases (i.e., como en las
ecuaciones 58 y 59). Para el intercambio Fe-Mg entre granate y biotita,
obtenenos a partir de (58):
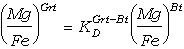 (62)
(62)
Esta relación
representa una recta en el espacio (Mg/Fe)Grt-(Mg/Fe)Bt,
cuya pendiente es igual a KD y su intersección en el origen es = 0
(Figura 6a). Por tanto, la proyección en un diagrama (Mg/Fe)Grt-(Mg/Fe)Bt
de rocas equilibradas a la misma presión y temperatura definirá una recta
si las fases se comportan como soluciones sólidas ideales, y la
proyección de rocas equilibradas a distinta temperatura o presiones definirá
distintas rectas. Relaciones lineales a P y T constantes también se obtienen en
diagramas ln(Mg/Fe)Grt-ln(Mg/Fe)Bt (Figura 6b). En
estos diagramas, la intersección para valores de la abcisa = 0 (i.e., (Mg/Fe)Bt=1)
corresponden a los valores de lnKD, como puede apreciarse tomando
logaritmos naturales en (62):
 (63)
(63)
Como hemos visto más arriba, la partición de
elementos entre dos fases es fuertemente dependiente de T, y en menor medida de
P. Como regla general, la partición decrece a medida que T aumenta, de manera
que el coeficiente de reparto KD (y Keq) tiende a 1, y lnKD
tiende a 0. Esto se debe a que la distinción energética entre los dos elementos
decrece con la temperatura, y las dos fases tienden a presentar razones
elementales más próximas (en el límite T=¥,
la razón elemental de las fases es idéntica e igual a la del sistema). En
consecuencia, la pendiente de las rectas en un diagrama (Mg/Fe)Grt-(Mg/Fe)Bt
tiende a 1 a medida que la temperatura de equilibramiento aumenta. En la
Figura 6a y b se han representado varias rectas correspondientes a distintos
valores de KD, menores de 1 para el caso en que la reacción de
intercambio se escriba como en (37), y mayores de 1 para el caso en que se
escriba en sentido contrario.
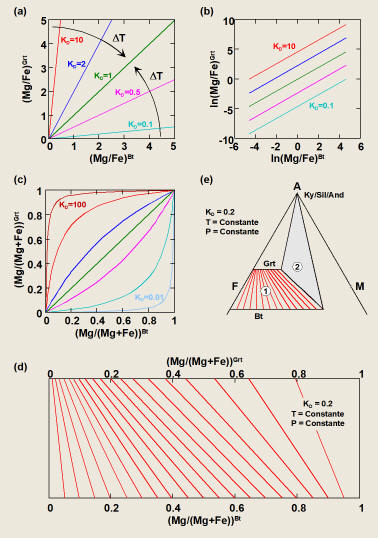
Figura 6.
Distintas formas de representar la fraccionación elemental entre dos fases,
ilustrada por el intercambio Fe-Mg entre granate y biotita.(a) y (b)
Diagramas (Mg/Fe)Grt-(Mg/Fe)Bt y ln(Mg/Fe)Grt-ln(Mg/Fe)Bt
para valores distintos de KD (i.e., a P y/o T variables). (c)
Diagrama de Roozeboom XMgGrt-XMgBt
para soluciones binarias Fe-Mg. (d) y (e) Diagrama de intercambio
recíproco y proyección AFM para condiciones P-T constantes bajo las cuales KD
es constante. Nótese que los valores de KD > 1 en las figuras (a),
(b) y (c) corresponden a la reacción de intercambio escrita en sentido contrario
a la ecuación (37).
Los diagramas representados en la Figura 6a
y b son de aplicación general ya que resultan de la definición general de KD.
Para el caso más restrictivo de soluciones binarias, la definición de KD
puede reescribirse a partir de la ecuación (58), teniendo en cuenta que XFea
= 1-XMga:
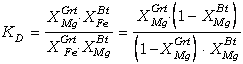 (64)
(64)
Reordenando, obtenemos:
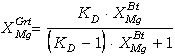 (65)
(65)
La representación
gráfica de la expresión (65) en un diagrama XMgGrt-XMgBt
(XMg=Mg/(Fe+Mg)) resulta en curvas cóncavas hacia arriba para KD<1
y cóncavas hacia abajo para KD>1, ambas simétricas respecto de la
diagonal ortogonal a la línea correspondiente a KD=1 (Figura 6c).
Estas curvas son las encontradas en el caso de partición entre soluciones
ideales (i.e., Kg=1).
Si una o las dos soluciones son no ideales, las relaciones de equilibrio
de intercambio también vendrán definidas por curvas, pero serán asimétricas
respecto de la diagonal anteriormente mencionada debido a que KD no
es constante a P y T fijas, sino que depende de la razón Mg/Fe de las fases
(i.e., de roca). Esto se debe a que Kg
depende de la composición (i.e., razón Mg/Fe) a P y T fijas cuando las
soluciones son no ideales, por lo que KD debe variar
proporcionalmente a Kg
para mantener Keq (= KD·Kg)
constante P y T fijas.
Las relaciones anteriores para mezclas binarias
pueden representarse también en diagramas de intercambio recíproco y en
proyecciones de fases apropiadas como la proyección AFM, (Figuras 6d y
e, respectivamente). Ambos tipos de diagramas son similares, ya que
representan la composición de fases coexistentes a P y T fijas mediante
líneas de compatibilidad. Sin embargo, en el primero de ellos no existen
limitaciones composicionales a la coexistencia de las dos fases (i.e., las
líneas de compatibilidad se distribuyen por todo el especto Mg/(Fe+Mg)roca).
Por el contrario, en los diagramas de fases de tipo AFM la composición de las
fases depende de las asociaciones de fases estables para distintas composiciones
de las rocas a P y T fijas. Así, rocas cuyas composiciones se proyecten en el
campo difásico Grt-Bt (campo 1 en la Figura 6e) presentarán composiciones de
granate y biotita variables en función de la composición de la roca, mientras
que rocas cuyas composiciones se proyecten en el campo trifásico Grt-Bt-Als
(campo 2 en la Figura 6e) presentarán composiciones de granate y biotita fijas,
independientemente de la composición de la roca (resultado de la regla de las
fases). Por otra parte, la proyección de fases coexistentes en rocas coincidirá
con las relaciones de partición descritas por la ecuaciones (64) y (65) e
ilustradas en las Figuras 6d y 6e si las soluciones son binarias e ideales. En
caso de soluciones no ideales, la distribución de las líneas de compatibilidad
será igualmente en forma de abanico, aunque KD no será constante a P
y T fijas, sino que variará con la composición del sistema por las mismas
razones que han sido expuestas anteriormente.
Para el caso más general de soluciones
multicomponentes, KD no puede ser definida como en la ecuación (64)
al ser una función de variables composicionales adicionales (e.g., XCaGrt;
XAl[VI],Bt,
además de XMgGrt y XMgBt) y por lo
tanto XMgGrt no queda definida de manera única para un
valor dado de XMgBt como en la ecuación (65). La
proyección de datos de partición entre fases de este tipo definirá también
curvas en diagramas XMgGrt - XMgBt,
cuya asimetría dependerá de las variaciones en otros componentes y del grado de
no idealidad de las soluciones. El efecto de componentes adicionales también
dependerá de la naturaleza de la no idealidad de la mezcla de estos elementos.
Como demostró el trabajo pionero de Kretz (1959), este efecto suele ser
sistemático, esto es, KD varia de forma sistemática con las
variaciones en los componentes adicionales. Por ejemplo, la mezcla de Ca y Mn en
granates ricos en Fe+Mg es no ideal, y es conocido el efecto de las variaciones
de estos elementos sobre el KD entre granate y biotita (Figura 7a):
a medida que Ca+Mn aumenta KD disminuye de forma lineal.
Esta dependencia lineal puede ser explicada por modelos de solución simétricos
para el granate, en combinación con interacciones energéticas casi ideales entre
Fe y Mg en las dos fases (e.g. Ganguly y Saxena, 1987, p.140).
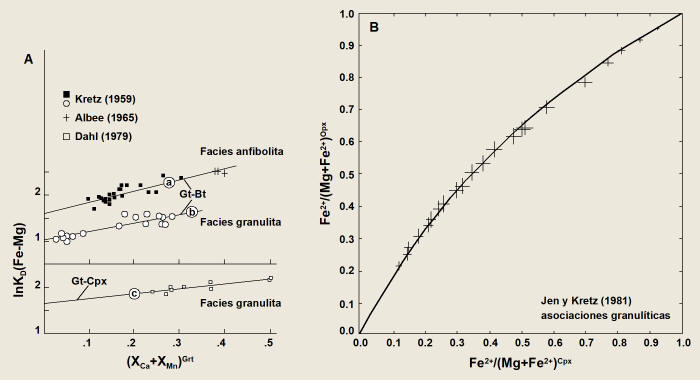
Figura 7.
a) Efecto sistemático de la variación en la concentración de Ca+Mn en el granate
sobre el KD(Fe-Mg)Grt-Bt/Cpx en rocas de grado medio y
alto; este efecto indica un comportamiento no ideal del granate (nótese que KD
= (Fe/Mg)Grt/(Fe/Mg)Bt/Cpx, i.e., el inverso del definido
a partir de la ecuación 37). b) Distribución Fe-Mg entre clinopiroxeno y
ortopiroxeno de asociaciones granulíticas (Jen y Kretz, 1981); el buen ajuste de
los datos a la curva sugiere que las rocas se equilibraron bajo condiciones P-T
similares. Figuras reproducidas de Ganguly y Saxena (1987).
La evaluación de las irregularidades en la
distribución de elementos entre fases coexistentes en las rocas, respecto de las
relaciones predecibles del comportamiento ideal, necesita considerar los efectos
de:
·
la proyección de muestras equilibradas a
distintas presiones y/o temperaturas,
·
la proyección de muestras con asociaciones
de fases en desequilibrio,
·
la dispersión analítica.
El análisis de estos puntos en series de rocas
puede llevarse a cabo si se dispone de modelos de solución que permitan calcular
Kg, ya que en
el equilibrio y a P y T fijas debe cumplirse que KD·Kg=constante.
Si un conjunto de rocas proyectadas en diagramas como los de la Figura 6 se
ajustan, dentro del error analítico, a líneas o curvas que cumplen la condición
KD·Kg=constante,
puede asegurarse que están equilibradas bajo rangos P-T suficientemente
restringidos (Figura 7b). En general, la dispersión analítica no puede
utilizarse como argumento para justificar los desajustes de las muestras a las
curvas calculadas bajo la condición KD·Kg=constante.
Si estos desajustes ocurren, es necesario concluir que los modelos de mezcla
utilizados no describen bien las soluciones implicadas en los equilibrios, que
las muestras no están equilibradas en rangos de P-T restringidos, o que las
muestras presentan asociaciones en desequilibrio.
Una evaluación cualitativa del grado en que las
rocas representan el equilibrio bajo condiciones de P y T fijas puede obtenerse
a partir de la observación de cruces de líneas de compatibilidad en diagramas de
fases del tipo AFM (y de intercambio recíproco), o, si las soluciones sólidas
que participan en los equilibrios son casi-ideales, a partir de los diagramas de
las Figuras 6a, 6b y 6c.
Los calibrados experimentales del equilibrio de
intercambio Fe-Mg entre biotita y granate han sido realizados por Ferry y Spear
(1978) y Perchuk y Laurent’eva (1983). Aunque existen dos calibrados empíricos
previos, propuestos por Thompson (1976) y Goldman y Albee (1977), es conveniente
comenzar la presente exposición por los calibrados experimentales,
particularizando en el realizado por Ferry y Spear (1978) ya que es el utilizado
más comunmente.
Ferry y Spear (1978) llevaron a cabo experimentos de equilibramiento de mezclas de
granate almandínico de composición conocida y constante (prp10alm90)
y biotita en la proporción 49:1 en el sistema pseudobinario Fe-Mg. Las
condiciones experimentales fueron isobáricas (2070 bar), a temperaturas entre
550 y 800 ºC. Debido a la elevada proporción de granate respecto de biotita, el
intercambio Fe-Mg entre las dos fases durante los experimentos no modifica de
forma efectiva la composición de granate, que permanece constante. Este diseño
condiciona que durante los experimentos sólo varie la composición de la biotita,
que en equilibrio es también rica en Fe. Así, la variación del coeficiente de
reparto KD observada en cada experimento, desde el inicio del mismo
(en desequilibrio, donde KD=KDdeseq) hasta su
final (en equilibrio aproximado, donde KD=KDeq),
se debe exclusivamente a las variaciones en la composición de la biotita. Los
experimentos de equilibramiento a cada presión y temperatura se realizaron de
forma que KDeq se aproximó desde los dos lados de la
reacción, esto es, partiendo de composiciones iniciales de biotita contrastadas
de forma que se obtuviesen valores de KDdeseq > KDeq
(i.e., en un lado de la reacción) y KDdeseq < KDeq
(en el lado opuesto).
Los resultados experimentales de Ferry y Spear
(1978) se presentan en la Figura 8a en un diagrama lnKD-1/T.
Este tipo de diagrama es conveniente para representar los resultados
experimentales isobáricos ya que, según las ecuaciones (35) y (41), lnKeq
es una función de 1/T a P constante. Si las soluciones sólidas son ideales (Kg=1;
Keq=KD) en el rango P-T-X de los experimentos y se
cumple que DCpr»0
(ec. 46) y DVro(P,T)ºconstante
(ec. 47), entonces los resultados experimentales deben ajustarse a una línea.
Como puede apreciarse en la Figura 8a, este es el procedimiento seguido
por Ferry y Spear (1978), que ajustan los datos experimentales a la recta:
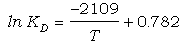 (66)
(66)
Comparando la ecuación (66) con la ecuación
(49), obtenemos:
DSro(1,298)/3·R
= 0.782 (67)
[-DHro(1,298)-P·DVro(1,298)]
/3·R = -2109 (68)
donde T es la
temperatura absoluta y el factor 3 aparece ya que la ecuación (49) está escrita
en función de Keq, y la reacción de intercambio se considera como en
la ecuación (37), de manera que Keq=KD3. De las
ecuaciones (67) y (68) pueden deducirse los incrementos de las propiedades
termodinámicas de la reacción de intercambio, teniendo en cuenta que la presión
de los experimentos es 2070 bar y que el incremento de volumen de la reacción
entre los componentes puros es 0.238 J/(bar·mol):
DSro(1,298)
= 3·R·0.782 = 19.51 J/(mol·K) (69)
DHro(1,298)
= 3·R·2109 - 2070·0.238 = 52112 J/mol (70)
(R=1.9872
cal/K·mol=8.3143 J/K·mol). Sustituyendo estos valores en la ecuación (49), el
calibrado de Ferry y Spear (1978) es:
0 = 52112 - 19.51·T(K)
+0.238·P(bar) + 3·R·T·lnKD (71)
que, despejando para T,
queda:
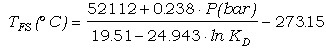 (72)
(72)
En la Figura 9 se presenta la solución
gráfica de este calibrado.
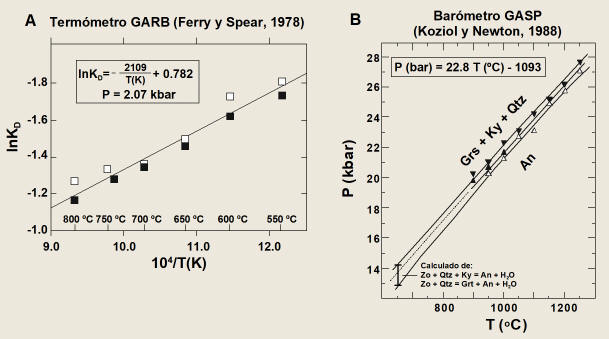
Figura 8.
Calibrados experimentales a) del termómetro GARB (Ferry y Spear, 1978) y b) del
barómetro GASP (Koziol y Newton, 1988).
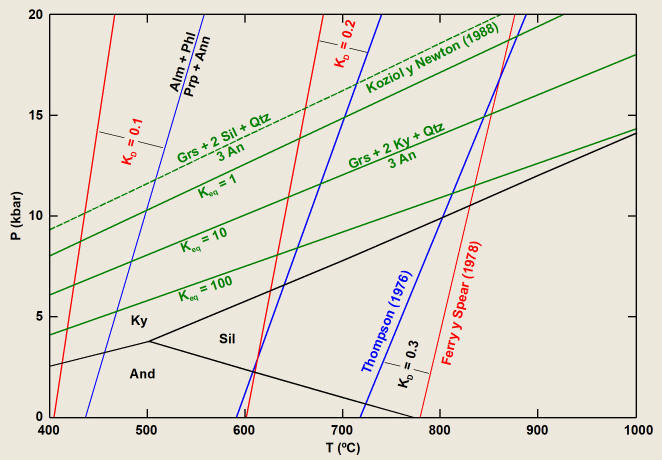
Figura 9.
Soluciones de algunos calibrados del termómetro GARB (Thompson, 1976, empírico;
Ferry y Spear, 1978, experimental) y barómetro GASP (Koziol y Newton, 1988;
experimental). Nótese que, por comparación con la Figura 5, en este caso se ha
representado KD en lugar de Keq (=KD3)
para el termómetro, y que la proyección del equilibrio GASP entre las fases
puras con silimanita es metaestable. Los campos de estabilidad de los polimorfos
de Al2SiO5 según Holdaway (1971).
Puede apreciarse que las isopletas de KD
son líneas con fuerte pendiente dP/dT. La función que define las pendiente es
dependiente del KD:
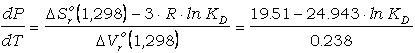 (73)
(73)
variando entre 323
bar/grado para KD=0.1 hasta 208 bar/grado para KD=0.3.
Esto implica que el termómetro es poco sensible a la presión de equilibramiento
asumida. Por otra parte, podemos apreciar la similitud de este calibrado con los
resultados presentados en la Figura 5, donde la ecuación que describe el
equilibrio no es lineal en P y T ya que incluye los incrementos de capacidad
calorífica, de expansión térmica y de compresibilidad.
La aplicación de la ecuación (72) a rocas
naturales debe ceñirse a las condiciones experimentales, i.e., entre 550 y 800
ºC, a bajas presiones y composiciones de granate y biotita dentro del sistema
pseudobinario Fe-Mg y con razones (Mg/Fe)roca bajas. Debido a la
escasa dependencia de P, las extrapolaciones en la presión asumida de
equilibramiento no son problemáticas. Las extrapolaciones en temperatura no son
problemáticas a alta T, ya que la coexistencia de granate+biotita en sistemas
ricos en Fe rara vez supera los 800 ºC. Las extrapolaciones a baja T son menos
fiables ya que no queda garantizada la dependencia lineal entre lnKD
y 1/T y las soluciones Fe-Mg se desvían más fuertemente de la idealidad. En este
sentido, parece que la suposición de comportamiento ideal de las soluciones en
el sistema pseudobinario Fe-Mg es apropiada para la biotita, aunque no para el
granate (e.g., Ganguly y Saxena, 1984, 1987).
La extrapolación en la composición de las fases
es, claramente, la fuente de errores más importantes en la estimación de la
temperatura de equilibramiento. Esto es debido al comportamiento no ideal de
mezclas cuaternarias Ca-Mn-Fe-Mg en el granate y Al-Ti-Fe-Mg (en las posiciones
octaédricas) en la biotita. Ferry y Spear (1978) recomiendan el uso de su
calibrado siempre que estos componentes adicionales se encuentre en baja
proporción, en concreto XCaGrt+XMnGrt<0.2
y XAl[VI],Bt+ XTi[VI],Bt<0.15.
Antes de considerar el efecto de otros
componentes, conviene describir brevemente el calibrado empírico de Thompson
(1976), ya que ilustra los métodos de calibración empírica más comunmente
utilizados.
Thompson (1976)
calibró el equilibrio de intercambio Fe-Mg entre biotita y granate a partir del
conocimiento del KD de asociaciones naturales cuyas temperaturas de
equilibramiento eran conocidas o asumidas por el autor (entre 500 y 700
ºC). La ecuación termométrica se obtiene mediante un análisis de regresión
lineal de lnKD sobre 1/T, que en este caso resulta en la ecuación
(r=0.976):
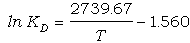 (74)
(74)
en que la reacción de
intercambio se escribe en sentido contrario al de la ecuación (37). Por esta
razón, los signos de las constantes n la ecuación (74) son los contrarios a los
encontrados por Ferry y Spear (1978), aunque los valores absolutos son similares
(cf. eq. 66).
Comparando las ecuaciones (74) y (49), y
teniendo en cuenta que Keq=KD3, se pueden
extraer los valores de las funciones termodinámicas molares mediante el
procedimiento seguido para el calibrado de Ferry y Spear (1978) anteriormente
discutido: DHr(1,298)=–68334.72
J/mol, DSr(1,298)=–38.11
J/mol·K y DVr(1,298)=–0.5837
J/bar·mol. Debe notarse que la aproximación empírica de Thompson (1976)
considera implícitamente los efectos de componentes adicionales en solución, y
las correspondientes desviaciones de la idealidad de las fases, ya que las fases
naturales analizadas contienen cantidades apreciables de otros componentes que
sustituyen a Fe y Mg. Por esta razón, los valores extraidos para los
incrementos de las propiedades termodinámicas no pueden hacerse equivalentes a
los correspondientes valores de reacción entre los componentes puros. En
sentido estricto, estos valores corresponden a los incrementos de las
propiedades termodinámicas de reacción entre los componentes puros más
los incrementos respectivos de las propiedades termodinámicas de exceso
de reacción. Sustituyendo los valores anteriores en la ecuación (49), y
dividiendo por 3·R para simplificar, obtenemos:
0 = -2739.646
+ 1.560·T -
0.0234·P + T· lnKD
(75)
que puede solucionarse
para T (P en bar):
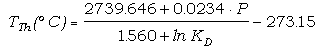 (76)
(76)
En la Figura 9 se presenta la solución
gráfica de este calibrado. En este caso, las isopletas de KD son
líneas con pendiente dP/dT menores que las correspondientes al calibrado de
Ferry y Spear (1978), debido a los distintos valores inferidos para los
incrementos de entropía y, sobre todo, de volúmen de reacción. La pendiente
dP/dT calculada a partir de:
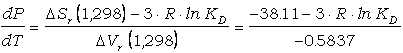 (77)
(77)
varía entre 164
bar/grado para KD=10 (equivalente a KD=0.1 para la
reacción escrita en sentido contrario, como en la ecuación 58) hasta 117
bar/grado para KD=3.33 (equivalente a KD=0.3 para la
reacción escrita en sentido contrario). Esto implica que este calibrado es más
sensible a la presión de equilibramiento asumida que el de Ferry y Spear (1978).
En principio, podría concluirse que el
calibrado de Thompson (1976) es más apropiado que el de Ferry y Spear (1978), ya
que implícitamente considera el efecto de componentes adicionales y las
desviaciones de la idealidad de las soluciones sólidas. No obstante, las
temperaturas calculadas con este termómetro llevan asociadas una elevada
incertidumbre debido a los errores en las estimaciones independientes de P y T
para las asociaciones que sirvieron para su calibrado. A pesar de ello, algunos
autores han señalado la validez “geológica” de este calibrado (e.g.,
Indares y Martignole, 1985, para rocas de alto grado).
Existen un gran número de modificaciones a los
calibrados precedentes que introducen correcciones a-X (modelos de actividad) a
fin de tener en cuenta las desviaciones de la idealidad de las soluciones
sólidas naturales. Estas correcciones se han implementado generalmente al
calibrado experimental de Ferry y Spear (1978), ya que las composiciones de las
fases utilizadas corresponden al sistema pseudobinario Fe-Mg, y por lo tanto en
los parámetros termodinámicos extraidos no existe término de exceso alguno
correspondiente a la mezcla de otros componentes. En otras palabras, si las
correcciones debidas a la mezcla no ideal de componentes adicionales son
necesarias, su formulación analítica debe implementarse sobre ecuaciones que
describan el sistema libre de esos componentes adicionales. Si estas
correcciones se aplicasen los calibrados de Thompson (1976) o Perchuk y
Laurent’eva (1983), que llevan implícitas estas correcciones, se sobrevaloraría
el efecto de mezcla no ideal de otros componentes.
Todos los estudios que han analizado el
intercambio Fe-Mg entre biotita y granate coinciden en la no-idealidad de ambas
fases (ver Ganguly y Saxena, 1987), aunque prácticamente todos ellos se han
focalizado en el problema de la solución sólida del granate, considerando que
gFe[VI],Bt=gMg[VI],Bt=1.
Para ilustrar los efectos de otros componentes, consideraremos aquí brevemente
algunas de las modificaciones del calibrado de Ferry y Spear (1978) más
populares.
Newton y Haselton (1981) y Hodges y Spear
(1982) consideran el efecto de la
desviación de la idealidad de las soluciones de granate y asumen que el
comportamiento de la biotita es ideal. En este caso, podemos expresar la
constante de equilibrio para el intercambio como (cf. ec. 55):
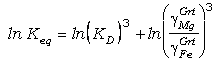 (78)
(78)
Estos autores modelizan el comportamiento no
ideal del granate mediante el modelo de solución cuaternaria simétrica de
Ganguly y Kennedy (1974) (o de mezcla simple, cf. Ganguly y Saxena, 1987,
p.135). Este modelo considera sólo las interacciones binarias y no las
multicomponentes en la expresión de la no-idealidad. Según este modelo, el
incremento de energía libre de exceso de la reacción (por mol de catión
intercambiado) es:
R·T·ln(gMg/gFe)Grt
= WFeMg(XFe–XMg) + (WMgCa–WFeCa)·XCa
+ (WMgMn–WFeMn)·XMn
(79)
donde Xi=i/(Fe+Mg+Ca+Mn)
y Wij son los parámetros de Margules de interacción binaria simétrica
i-j en el sistema cuaternario Ca-Mn-Fe-Mg. Este modelo no es aceptado
actualmente debido, sobre todo, a la suposición de interacciones binarias
simétricas. Newton y Haselton (1981) y Hodges y Spear (1982) consideran todos
los parámetros de Margules = 0 excepto el parámetro de interacción entre Mg y
Ca:
WMgCa
= 3300 – 1.5·T cal/mol de catión = 13807.2 – 6.276·T J/mol de catión
(80)
Sustituyendo (80) y
(79) en (78), y sustituyendo la expresión resultante en la ecuación (55), junto
con los incrementos de las propiedades termodinámicas de la reacción de
intercambio extraídos del calibrado de Ferry y Spear (1978), obtenemos la
corrección siguiente:
0 = 12454 - 4.662·T +
0.057·P + 3·R·T·lnKD +
3·(3300 - 1.5·T)·XCaGrt (81)
y despejando para T:
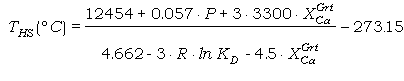 (82)
(82)
donde las constantes se
han expresado en calorías en lugar de en julios ya que es la formulación común
en la literatura.
Ganguly y Saxena (1984, 1985),
tras un análisis de los datos termodinámicos disponibles sugieren que la mezcla
en el sistema binario Fe-Mg es asimétrica, y ofrecen otros valores para los
parámetros de Margules de interacción binaria:
WMgCa–WFeCa =
DWCa
= 3000 ± 500 cal/mol de catión (83)
WMgMn–WFeMn
=
DWMn
= 3000 ± 500 cal/mol de catión (84)
WFeMg =
WFe-Mg· Mg/(Mg+Fe) + WMg-Fe·(Fe/(Mg+Fe)
(85)
WFe-Mg
= 200 cal/mol de catión (86)
WMg-Fe = 2500 ± 500 cal/mol de catión
(87)
La corrección
resultante es:
0 = 12454 –
4.662·T + 0.057·P + 3·R·T·lnKD +
3·[WFeMg·(XFeGrt–XMgGrt)
+
DWCa·XCa
+
DWMn·XMn]
(88)
de la que hay que
despejar para T (con alguna otra modificación, ver Ganguly y Saxena, 1984 y 1985
para más detalles). Los parámetros de mezcla, y por lo tanto la corrección del
calibrado, están definidos para composiciones de XCaGrt y
XMnGrt<0.3, y para valores de (Fe/Mg)Grt>3; en
caso contrario la suposición de mezcla asimétrica no ideal entre Fe-Mg no está
garantizada, por lo que podría obviarse el parámetro WFeMg en la
formulación termométrica (como es el caso de granates ricos en prp de grado
alto).
Existen otras correcciones más recientes
basadas en modelos de solución más refinados para el granate, entre los que
pueden citarse los de Anovitz y Essene (1987), Geiger et al. (1987), Koziol y
Newton (1989), Koziol (1990) y Berman (1990). En el caso de Berman (1990),
la solución se modeliza mediante un modelo general asimétrico cuaternario, y la
ecuación que define el coeficiente de actividad del componente m es un polinomio
de tercer grado en la composición expresable como:
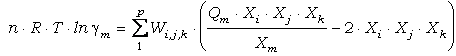 (89)
(89)
donde cada parámetro de Margules Wi,j,k
es una función de P y T: WGi,j,k=WHi,j,k-T·WSi,j,k+P·WVi,j,k
(WH, WS y WV se asumen constantes
independientes de P, T y la composición), Qm es un término que da
cuenta del número de i, j y k que son iguales a m, y p es el número de
parámetros de mezcla necesarios para una solución dada (número de permutaciones
posibles entre i, j y k). Berman (1990) calculó los parámetros ternarios a
partir de las interacciones binarias según el modelo de Wohl (1953):
Wi,j,k = (Wi,i,j + Wi,j,j + Wi,i,k
+ Wi,k,k + Wj,j,k + Wj,k,k)/2 - Ci,j,k
(90)
aunque haciendo el
término ternario Ci,j,k = 0 debido a la ausencia de datos
experimentales necesarios para calibrar las interacciones ternarias. La
extensión de la ecuación para almandino, por ejemplo, es:
3·R·T·lngFeGrt
= W112(-2X12X2) + W122(-2X1X22) + W113(X12-2X12X3)
+ W133(2X1X3-2X1X32) +
+W114(-2X12X4) + W144(-2X1X42) + W223(X22-2X22X3) + W233(2X2X3-2X2X32) +
+W224(-2X22X4) + W244(-2X2X42) + W334(2X3X4-2X32X4) + W344(X42-2X3X42) +
+ W123(X1X2-2X1X2X3) + W124(-2X1X2X4)+
+ W134(X1X4-2X1X2X4) + W234(X2X4-2X2X3X4)
(91)
donde las Xi
se refieren a las fracciones molares de 1=Ca, 2=Mg, 3=Fe, 4=Mn, y Wi,j,k
como en la ecuación (89). La expresión de
gFeGrt
(91) y la correspondiente para gMgGrt
deben sustituirse en la ecuación (78), y ésta última a su vez debe sustituirse
en la ecuación (55), junto con los incrementos de las propiedades termodinámicas
de la reacción de intercambio extraidos del calibrado de Ferry y Spear (1978)
para obtener la corrección al termómetro. Nótese que en este caso, los Wi,j,k
son funciones de P, T y Xi, y el despeje de T es algo más complicado
que en los casos anteriores. En principio, esta corrección permite extrapolar el
calibrado de Ferry y Spear a cualquier composición de granate.
De la misma manera que para el granate, se han
desarrollado expresiones de los coeficientes de actividad para los componentes
en solución en la biotita. Estas han sido propuestas a partir del análisis de
datos naturales (Indares y Martignole, 1985; Sengupta et al., 1990; Hoisch,
1991), y experimentales (Patiño Douce et al., 1993). Como ejemplo,
consideraremos aquí la corrección de Indares y Martignole (1985)
Estos autores consideran el efecto del Ti y Al[VI]
en la desviación de la idealidad de la biotita, asumiendo un modelo de solución
cuaternaria simétrica Fe-Mg-Ti-Al en las posiciones octaédricas (Ganguly y
Kennedy, 1974). El cálculo de DWAl[VI],Bt=WMg-Al–WFe-Al
y DWTiBt=WMg-Ti–WFe-Ti)
es empírico, realizado por análisis de regresión sobre muestras de granulitas
donde existen estimaciones independientes de P y T. El método es similar al
utilizado por Thompson (1976), aunque en este caso la ecuación a la que se
ajusta la regresión contiene como incógnitas los parámetros anteriores (DWAl[VI],Bt=m,
y DWTiBt=n)
y como coeficientes conocidos los incrementos de las propiedades termodinámicas
de la reacción entre los componentes puros y los parámetros de mezcla del
granate. En base a estos últimos, desarrollan dos formulaciones distintas,
siguiendo los modelos de Newton y Haselton (1981) y Hodges y Spear (1982) por
una parte (modelo A), y de Ganguly y Saxena (1984) por otra (modelo B). Este
último modelo es modificado por Indares y Martignole (1985), quienes sugieren
que el parámetro de Margules que describe la no-idealidad de la solución Fe-Mg
(i.e. WFeMg) en el granate puede no ser válido para valores de
(Fe/Mg)Grt<3, tal y como señalan Ganguly y Saxena (1984), y para
condiciones de alta T, por lo que no incluyen este parámetro en la regresión de
los datos para obtener su corrección B. Las ecuaciones termométricas son (los
parámetros y constantes en calorías):
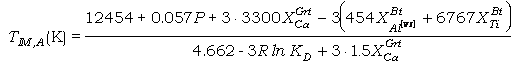 (92)
(92)
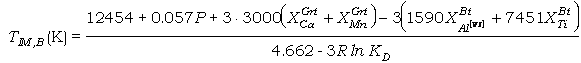 (93)
(93)
donde los parámetros
DWAl[VI],Bt
y DWTiBt
obtenidos en las regresiones son los coeficientes que multiplican a XAl[VI],Bt
y XTiBt, respectivamente, en cada ecuación.
Algunos autores han señalado los buenos
resultados obtenidos con estas correcciones (e.g., Bohlen y Lindsley, 1987),
aunque existen bastantes limitaciones en su aplicabilidad. En primer lugar, debe
restringirse a condiciones de facies de granulitas. Por otra parte, las
correcciones propuestas pueden no ser satisfactorias para describir los efectos
del Al y Ti en las biotitas, ya que el rango de variación composicional de las
biotitas usadas por Indares y Martignole (1985) en las regresiones es muy
limitado para resultar en valores de
DWAl[VI],Bt
y DWTiBt
significativos (ver Ganguly y Saxena, 1987, p 238). Así, Sengupta et al. (1990)
han evaluado los parámetros (WMg-Al–WFe-Al)Bt y
(WMg-Ti–WFe-Ti)Bt para el intercambio Fe-Mg
entre ortopiroxeno y biotita en granulitas siguiendo la misma técnica que
Indares y Martignole (1985), resultando en 3595 cal/mol catión y 4423 cal/mol
catión, respectivamente. Estos valores contrastan con los ofrecidos por Indares
y Martignole (1985), sobre todo para el caso del modelo A (ver también Hoisch,
1991 y Patiño Douce et al., 1993). Finalmente, las restricciones impuestas por
las muestras naturales utilizadas en las regresiones implican que estos
calibrados deben aplicarse a rocas en las que (Fe/Mg)Grt<3 y donde el
balance del Al[VI] y Ti en la biotita se verifique mediante la
sustitución Ti-vacante y di-trioctaédrica (este último aspecto es remarcado por
Indares y Martignole, 1985).
A pesar de estas limitaciones, muchos autores
han aplicado los calibrados de Indares y Martignole (1985) bajo condiciones
P-T-X fuera del rango utilizado en los calibrados, como en rocas de grado medio
donde (Fe/Mg)Grt > 3, y sin demostrar que la solución de biotita en
las posiciones octaédricas se ajusta a las premisas indicadas más arriba.
Como se ha indicado más arriba, los equilibrios
de transferencia neta sólido-sólido suelen presentar incrementos de volumen de
reacción suficientes para ser utilizados como barómetros (Tabla 3). De entre
ellos, el basado en el equilibrio entre granate, silicato de Al, cuarzo y
plagioclasa (GASP), expresado mediante la reacción de transferencia neta entre
los componentes de fase grosularia, silicato de Al, cuarzo, y anortita (ec. 36;
ver Tabla 3 para referencias), ha sido uno de los mejor estudiados.
El calibrado de este equilibrio puede abordarse
partiendo de la condición (23):
 (94)
(94)
donde las actividades
de los componentes cuarzo y silicato de Al son aqtzQtz=aalsKy,Sil,And=1
ya que las fases cuarzo y silicato de Al pueden considerarse puras. La
aplicación práctica de este barómetro necesita, por tanto, conocer formulaciones
explícitas para DGro(P,T)
y disponer de modelos de actividad para el granate y la plagioclasa. Abordaremos
estos dos aspectos a continuación.
El calibrado del equilibrio entre las fases
puras es el primer estadio en la calibración de una reacción, ya que permiten
deducir los incrementos de las propiedades termodinámicas que permite conocer
DGro(P,T).
La reacción entre las fases puras define el límite superior de estabilidad de
anortita a alta presión, y su localización en el espacio P-T tiene lugar en el
campo de estabilidad de la distena (Figura 8b).
Las calibraciones experimentales de la reacción
GASP entre las fases puras incluyen los trabajos de Hays (1967), Hariya y
Kennedy (1968), Schmid et al. (1978), Goldsmith (1980), Gasparik (1984) y Koziol
y Newton (1988). De ellos, el calibrado de Goldsmith (1980) primero, y el de
Koziol y Newton (1988) después, han sido los más utilizados para realizar
estimaciones de presión. A partir de los datos de Goldsmith (1980), la
ecuación que describe la reacción entre las fases puras con distena es:
PO
(bar) = -2100 + 23.2·T (ºC), con distena (95)
De este calibrado y del
calibrado experimental de la reacción Ky=Sil de Holdaway (1971), Newton y
Haselton (1981) ofrecen la correspondiente ecuación para la reaccion con
silimanita:
PO (bar) = -600 + 23.6·T (ºC),
con silimanita (96)
Los experimentos de Goldsmith (1980) fueron
conducidos a alta T (>900 ºC) por necesidades cinéticas,
persiguiendo acotar de manera lo más precisa posible el equilibrio mediante
inversiones en la dirección de la reacción. En las aplicaciones prácticas del barómentro GASP, esto introduce una fuente incertidumbre adicional a la del
propio calibrado experimental, ya que hay que hacer una extrapolación a
temperaturas propias del metamorfismo, generalmente menores de 800 ºC. Debido a
esta fuente de error, se reconoce una incertidumbre de ±3 kbar para las
presiones calculadas con este calibrado.
Más recientemente, Koziol y Newton (1988) han
calibrado la reacción entre las fases puras contando con mejoras técnicas que
permiten reducir el error experimental. Además, aunque los experimentos se
realizaron a alta P y T (>900 ºC y >18 kbar), estos autores localizan un punto
de la reacción a baja P y T (650 ºC
y entre 14-15 kbar, obtenido a partir de los calibrados experimentales de las
reacciones 2Zo+Ky+Qz=4An+H2O
y 4Zo+Qz=5An+Grs+2H2O,
Figura 8b) que permite reducir sustancialmente la incertidumbre debida a
la extrapolación de la reacción entre las fases puras al rango P-T propio del
metamorfismo cortical. La incertidumbre asignada por Koziol y Newton (1988) es
de ±650 bar a 600
ºC y ±400 bar a 1000 ºC.
Los calibrados de la reacción entre las fases puras con distena y silimanita que
ofrecen estos autores son (Figuras 8b y 9):
PO
(bar) = -1093 + 22.80·T (ºC) con distena (97)
PO (bar) = -25 + 23.41·T (oC)
- 0.0001872·T2 (ºC) con silimanita (98)
En la Figura 9 puede apreciarse que el
equilibrio estable entre las fases puras ocurre en el campo de la distena, y,
consecuentemente, el equilibrio con silimanita es metaestable. A partir de las
ecuaciones (97) y (98) pueden extraerse los incrementos de las propiedades
termodinámicas de las reacciones entre las fases puras. El procedimiento es
idéntico al realizado anteriormente para los calibrados del termómetro
biotita-granate, esto es, se asume que
DCpr = 0 y
DVr(P,T) =
constante. Como ejemplo, analizaremos la reacción con distena. En primer lugar
hay que transformar la ecuación (97) para expresar T en grados Kelvin;
despejando T (ºC) en (97), sumando 273.15 ºC y volviendo a despejar para P
obtenemos:
POky (bar) = -7320.8 + 22.80·T (K)
(97b)
Considerando que el
incremento de volúmen para la reacción con distena es
DVro(1,298)=6.605
J/mol·bar (calculado a partir de los datos termodinámicos de Holland y Powell,
1990), y teniendo en cuenta que en el equilibrio entre los componentes puros Keq=1
(i.e., R·T·lnKeq=0), tenemos, reordenando (97b) y multiplicando por
DVro(1,298):
0 = (6.605)·7320.8 - (6.605)·22.80·T + (6.605)·Poky
+ R·T·ln(1)
0 = 48353.9 - 150.69·T + 6.605·POky
(99)
de donde se deduce que
DHro(1,298)=48353.9
J/mol y DSro(1,298)=150.69
J/mol·K. Sustituyendo estos valores en la ecuación (49) se obtiene la expresión
barométrica:
0 = 48353.9 -150.69·T (K) + 6.605·P + R·T·lnKeq
(100)
que puede despejarse
para P si las funciones explícitas de las actividades de grosularia y anortita
no dependen de P (lo cual no es el caso, como veremos más adelante). La solución
gráfica de este calibrado se presenta en la Figura 9. Puede apreciarse
que las isopletas de Keq son lineas con pendiente dP/dT baja:
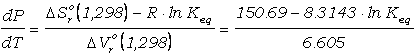 (101)
(101)
que varía de 22.8
bar/grado para Keq=1 hasta 11.2 bar/grado para Keq=10000
(en este caso el equilibrio con distena es metaestable, como se ilustra en la
Figura 5, por lo que no ha sido representado en la Figura 9). Un
procedimiento idéntico puede seguirse para las reacciones con silimanita y
andalucita (ver Figura 5, donde la función que describe el equilibrio no
es lineal en P y T ya que incluye los incrementos de capacidad calorífica,
expansión térmica y compresibilidad isoterma).
De la ecuación (100) puede deducirse que el
aumento de presión (a T constante) implica disminuir el valor de Keq
(Figuras 5 y 9), lo que supone aumentar los valores XCaGrt,
disminuir Xan, o ambos. Esto es lo que ocurre en las rocas naturales,
donde se observa que a medida que aumenta la presión de equilibramiento, el
granate es más rico en grosularia y la plagioclasa en albita. No obstante, este
criterio cualitativo no es aplicable en sentido estricto ya que, en sistemas
naturales multivariantes la composición de las fases es dependiente de P, T y de
la composición del sistema. Así, rocas ricas en Ca presentarán granate y
plagioclasa ricos en Ca, y es posible encontrar rocas equilibradas bajo las
mismas condiciones P-T con composiciones de las fases contrastadas, aunque la
constante de equilibrio debe ser la misma.
Ghent (1976)
mostró la aplicabilidad del barómetro GASP en asociaciones metapelíticas de
medio y alto grado, si bien, ante la ausencia de modelos de solución asequibles
para el granate y plagioclasa consideró un comportamiento ideal para ambas fases
(i.e., agrs=XCaGrt y aan=Xan).
Este autor indicó que los datos experimentales obtenidos por Ganguly y Kennedy
(1974) en granates naturales y por Orville (1972) en plagioclasa indican que las
actividades de grosularia en concentraciones diluidas y de anortita y albita son
mayores que sus fracciones molares. Aunque la no consideración de estos
comportamientos no ideales supone infraestimar las estimaciones de P en el
primer caso (granate) y sobreestimarlas en el segundo (plagioclasa), Ghent
(1976) sugirió que ambos efectos parecen cancelarse mutuamente, por lo que las
estimaciones realizadas por Ghent (1976) considerando las soluciones ideales en
distintas asociaciones naturales coinciden aproximadamente con las predecibles a
partir de los campos de estabilidad de los polimorfos de Al2SiO5.
Con posterioridad a Ghent (1976), la mayoría de las aplicaciones de este
barómetro han considerado modelos de mezcla no ideales.
La solución sólida de granate suele describirse
mediante modelos de mezcla generales, de tipo iónico multiposicional, como los
presentados más arriba (ec. 53). En estos modelos, la función de actividad de la
grosularia que debe sustituirse en la ecuación (94) es:
agrs=Xgrs3·ggrs3
(102)
ya que la multiplicidad
de la posición estructural donde se mezcla el Ca es 3, según se ha definido la
reacción en la ecuación (36).
Lo mismo puede decirse de la solución sólida de
plagioclasa, aunque en este caso existen complicaciones adicionales derivadas de
su estado de ordenamiento y del principio de Al-avoidance. Según este
principio, la mezcla en las posiciones tetraédricas no es completamente azarosa,
de forma que que la estructura de la plagioclasa no presentaría enlaces Al-O-Al
encadenados, lo cual supone modificar la expresión de la entropía de mezcla
iónica ideal (Kerrick y Darken, 1975; Tema 3). Ejemplos de modelos de soluciones
binaria (ab-or) y ternaria (ab-an-or) son Saxena y Ribbe (1972), Newton et al.
(1980), Newton y Haselon (1981), Hodges y Spear (1982), Ghiorso (1984), Green y
Usdansky (1986), Fuhrman y Lindsley (1988) y Elkins y Grove (1990). En el caso
del modelo de Fuhrman y Lindsley (1988), la expresión de aan es:
aan =
[Xan·(1 + Xan)2/4]· exp{(Wor-ab[XabXor(0.5-Xan-2Xab)] + Wab-or[XabXor(0.5-Xan-2Xor)]
+
+ Wor-an[2XorXan(1-Xan)+XabXor(0.5-Xan)] + Wan-or[Xor2(1-2Xan)+XabXor(0.5-Xan)]
+
+ Wab-an[2XabXan(1-Xan) + XabXor(0.5-Xan)] + Wan-ab[Xab2(1-2Xan) + XabXor(0.5-Xan)]
+
+ Wor-ab-an[XorXab(1-2Xan)])/(R·T)}
(103)
donde los valores de
los parámetros de Margules W son funciones de P y T: WG=WH-T·WS+P·WV
(WH, WS y WV se asumen constantes
independientes de P, T y la composición). Como puede apreciarse, en este modelo
de mezcla las relaciones a-X no son del tipo ai=Xi·gi.
Esto se debe a que el término ideal de la actividad de la anortita, que
es el primer factor del lado derecho de la expresión que multiplica al
exponencial (i.e., al coeficiente de actividad), resulta del principio de Al-avoidance
mencionado anteriormente.
La utilización de modelos de mezcla como el
anterior, donde la actividad de los componentes depende explícitamente de P y T,
hace necesario despejar P de la función de Keq para obtener una
solución de presión.
Notas
Por
simplicidad se omite la consideración de las interacciones energéticas
recíprocas (i.e., interposicionales), que dan lugar al coeficiente de
actividad recíproco gka,
que multiplica al producto de la ecuación (52).
T1
y T2 representan dos posiciones tetraédricas distinguibles en la
biotita, una con multiplicidad 1 ocupada por Al y otra con multiplicidad
3 ocupada por silicio. La distinción de estas posiciones hace que el
producto Õ(Xigk)Bt
deba ser multiplicado por el factor corrector 1/[(0.25)·(0.75)3],
necesario para que la actividad de la annita pura sea igual a 1 (Tema 3
del programa de Petrogénesis Metamórfica I. Aproximación Termodinámica).
Nótese
que al reescribir las reacciones de intercambio en términos de un mol de
ión intercambiado se modifica la función Keq y también el
incremento de energía libre de reacción entre los componentes
moleculares puros DGro(P,T),
ya que las funciones de estado (i.e., energía libre, entalpía, entropía,
volúmen, etc) de cada componente molecular deben multiplicarse por sus
respectivos coeficientes estequiométricos (i.e., 1/3 para almandino y
piropo y 1 para diópsido y hedenbergita en el caso del equilibrio Fe-Mg
entre granate y clinopiroxeno.
Valores
de KD < 1 y KD > 1 (i.e., cambios en el signo de
lnKD) para una reacción de intercambio escrita de forma única
pueden darse si el sistema es azeotrópico (Tema 3), resultado del
comportamiento no ideal de una o las dos fases.
Nótese
que en la Figura 7, tomada de Kretz, 1959, KD está definido a
partir de la reacción de intercambio escrita en sentido contrario a la
ecuación (37).
La
ecuación correspondiente al calibrado experimental de Perchuk y
Larent’eva (1983) es: lnKD = 3947.5/T-2.868. Estos autores
utilizaron fases naturales desviadas del sistema pseudobinario Fe-Mg,
por lo que las estimaciones de T llevan implícitas desviaciones de la
idealidad debidas a la presencia de otros componentes, aunque debe
tenerse en cuenta que las cantidades de Ca y Mn de los granates usados
por estos autores en los experimentos son bajas.
A
partir de una comunicación personal de Newton, Ganguly y Saxena (1984)
señalan que esta ecuación es errónea; la expresión que debe utilizarse
para el equilibrio con silimanita es (derivada por E. Froese): Po
(kbar) = -1.17 + 0.0238·T (C).
[Back] [Home] [Up] [Next]
Última
modificación:
domingo, 05 de octubre de 2025 15:27 +0200 |
