3.1 Aplicaciones de los biochips
![]()
Laboratorio de ADN forense, Depto. de Medicina Legal
Universidad de Granada, España
1. Evolución de las técnicas de estudio de los polimorfismos de ADN
2. Análisis por la reacción en cadena de la polimerasa (PCR)
3. Futuras tecnologías: biochips
3.1 Aplicaciones de los biochips
La identificación con ADN o “huella genética” se basa en el estudio de una serie de fragmentos de ADN presentes en todos los individuos pero que poseen la característica de ser altamente variables o polimórficos entre los mismos.
El análisis de un determinado número de estas secuencias o fragmentos de ADN permite identificar a un individuo con una probabilidad muy cercana al 100%.
Además de ser muy polimórfico, el ADN que se utiliza para la identificación en Genética Forense es un ADN no codificante o no expresivo, por lo que no revela características fenotípicas de los individuos; este hecho es de gran importancia a la hora de considerar la creación de las bases de datos genéticas.
Para analizar dichos polimorfismos del ADN, los laboratorios de Genética Forense utilizan una serie de técnicas que están en continua evolución, consiguiendo que cada vez la identificación por medio del ADN sea más precisa y rápida.
![]()
La Hemogenética Forense nace a principios de siglo, cuando Karl Landsteiner describe el sistema ABO de los hematíes y Von Durgen y Hirschfeld descubren su transmisión hereditaria. Esta ciencia surgió como una rama de la Criminalística cuyo objetivo era la identificación genética tanto en casos de investigación criminal como en estudios biológicos de la paternidad. Inicialmente, las investigaciones se centraban en el estudio de antígenos eritrocitarios (sistema ABO, Rh, MN), proteínas séricas, enzimas eritrocitarias y sistema HLA. Con el estudio de dichos marcadores podía incluirse o excluirse una persona como posible sospechoso por poseer una combinación genética igual o diferente a la del vestigio biológico hallado en el lugar de los hechos.
Pero fue a mediados de siglo cuando gracias al descubrimiento del ADN y de su estructura y al posterior avance en las técnicas de análisis de dicha molécula la Hemogenética Forense evolucionó considerablemente hasta el punto de que hoy en día puede hablarse de una nueva subespecialidad dentro de la Medicina Forense: la Genética Forense. Dicha ciencia estudia básicamente unas regiones del ADN que presentan variabilidad entre los distintos individuos, es decir, estudia regiones polimórficas del ADN. Así, analizando un determinado número de regiones polimórficas, la probabilidad de que dos individuos sean genéticamente iguales es prácticamente nula (excepto en el caso de gemelos univitelinos).
Aunque la Ciencia poseía las herramientas necesarias para el estudio del ADN, su aplicación en la resolución de casos judiciales no se produjo hasta 1985, cuando el Ministerio del Interior Británico solicitó la ayuda de Alec J. Jeffreys, profesor de Genética de la Universidad de Leicester. Los primeros casos de Criminalística fueron resueltos gracias a la técnica de los RFLPs (Fragmentos de Restricción de Longitud Polimórfica). Jeffreys descubrió la existencia de unas regiones minisatélites hipervariables dispersas por el genoma humano que al ser tratadas con enzimas de restricción generaban fragmentos de longitud variable. Estudios posteriores realizados el mismo Jeffreys demostraron que las diferencias en el tamaño de estos fragmentos se debían a que estas regiones consistían en un determinado número de repeticiones en tándem de una secuencia central, el cual variaba de unos individuos a otros.
El primer locus de ADN polimórfico fué descubierto por Wyman y White en 1980 usando una sonda de ADN arbitraria. De esta manera observaron fragmentos de más de 15 longitudes diferentes en una pequeña muestra de individuos. Posteriormente se encontraron otros loci hipervariables como en la secuencia del gen de la insulina humana, en el oncogen “ras”, en el pseudogen de la zeta-globina y en el gen de la mioglobina. Estos loci hipervariables constaban de repeticiones en tándem de una secuencia de oligonucleótidos (11 a 60 pb), de manera que las diferentes longitudes de los fragmentos originados dependían del número de dichas repeticiones y se les denominó VNTR (“Variable Number of Tandem Repeat”).
Tras el descubrimiento de los primeros VNTRs se vio que éstos podían ser aplicados a la medicina forense y sustituir a los marcadores clásicos.
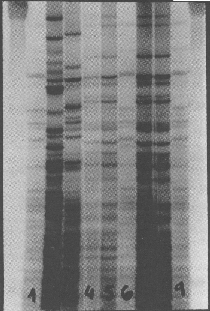
En un principio la manera de estudiar dichos marcadores se hizo por medio de la técnica llamada hibridación con sondas o Southern blot. Esta técnica consta básicamente de las siguientes etapas:
Digestión del ADN con enzimas de restricción tras conseguir extraer un ADN de alta molecularidad.
Separación de los fragmentos obtenidos por medio de una electroforesis en gel de agarosa.
Desnaturalización de los fragmentos separados y cortados.
Transferencia de las cadenas simples a una membrana de nitrocelulosa o nylon y fijación de las mismas por medio de calor (80ºC).
Prehibridación con sondas de ADN inespecífico para bloquear los lugares de unión inespecíficos que pudiera haber en la membrana.
Marcaje de la sonda con nucleótidos radioactivos (32 P normalmente).
Hibridación de la sonda marcada y desnaturalizada con los fragmentos de ADN fijados a la membrana, y lavado de la membrana para eliminar el exceso de sonda o aquellas que hayan hibridado mal.
Revelado en placa radiográfica e interpretación de los resultados.

El tipo de sondas utilizadas puede ser de dos tipos:
|
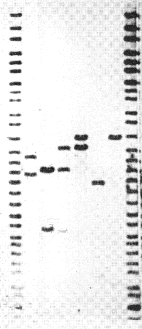
Sondas Mono-locus (SLP): son específicas para una región de un determinado cromosoma. Se unen a secuencias largas de nucleótidos y presentan mayor variabilidad que las sondas multi-locus. Como resultado se observan una o dos bandas por individuo, según sea homocigoto o heterocigoto. El patrón de bandas obtenido con estas sondas se denomina perfil unilocus de ADN o “DNA profiling” | |
|
Sondas Multi-locus (MLP): hibridan con secuencias minisatélites presentes en varios loci de diferentes cromosomas. Son sondas de 10 a 15 nucleótidos que se repiten múltiples veces y tras el revelado se observan de 10 a 20 bandas por persona. Este patrón de múltiples bandas se conoce como huella genética multilocus o “DNA fingerprint”. |
| Sondas multilocus | Sondas unilocus |
|
|
 |
Las sondas multi y mono-locus presentan una serie de ventajas e inconvenientes con respecto a una serie de parámetros como son:
|
Información aportada: las sondas multi-locus tienen una mayor capacidad discriminativa al aparecer múltiples bandas. No obstante, las mono-locus son más específicas ya que el fragmento de ADN con el que hibridan es de mayor tamaño. | |
|
Cantidad y calidad del ADN: cuando se usan sondas multi-locus se requiere aproximadamente un microgramo de ADN sin degradar mientras que en el caso de las mono-locus se necesita menos de 100 ng y este ADN no necesariamente debe estar en perfecto estado, siempre y cuando el fragmento complementario a la sonda esté intacto. | |
|
Especificidad entre especies: las sondas multi-locus permiten su uso sobre el ADN humano y de cientos de animales superiores, mientras que las mono-locus son exclusivas de ADN humano. |
A pesar de que el análisis SLP ha sido y es bastante útil en estudios de paternidad no puede decirse lo mismo de su aplicación a la Criminalística ya que presenta una serie de inconvenientes como son:
|
La cantidad de ADN que se necesita está entre 20 y 100 ng, cantidad difícil de conseguir en casos de criminalística en los que los indicios biológicos encontrados son mínimos. | |
|
En cuanto a la calidad del ADN, en la práctica forense es muy difícil encontrar en estado no degradado toda la cantidad de ADN que se necesita para un análisis con sondas mono-locus. | |
|
El tiempo requerido para este tipo de análisis es de dos o tres días. | |
|
El hecho de que se requieran cantidades elevadas de ADN hacen que normalmente, con el primer análisis se consume la totalidad de la muestra, con lo que se dificultan contrapericias y una posterior revisión del caso. |
Todas estas limitaciones fueron superadas gracias a la aplicación en Genética Forense de una técnica, la Reacción en Cadena de la Polimerasa (“PCR”), que supuso una revolución en muchos campos de la Biología y de la Medicina.
El estudio de indicios biológicos por PCR ha permitido la resolución de un gran número de casos en Criminalística que hasta entonces eran desestimados por no poseer la suficiente cantidad de muestra para su análisis por RFLP. Con el uso de la PCR muestras tan mínimas como pueden ser un pelo con raíz, una minúscula mancha de sangre o semen e incluso caspa son suficientes en muchos casos para llevar a cabo un análisis de identificación genética.
![]()
La reacción en cadena de la polimerasa (conocida como PCR por sus siglas en inglés, Polymerase Chain Reaction) permite amplificar más de un millón de veces un ADN obtenido a partir de una región seleccionada del genoma, siempre y cuando se conozca una parte de su secuencia de nucleótidos. Esta técnica fue ideada en 1989 por Kary B. Mullis que obtuvo el premio Nobel de Química en 1993 por dicho invento.
Para la PCR se utilizan dos oligonucleótidos sintéticos de unos 15-20 nucleótidos que son complementarios a las zonas flanqueantes de la región que se quiere amplificar. Estos oligonucleótidos (habitualmente conocidos por su nombre en inglés, "primers") actúan como cebadores para la síntesis in vitro de ADN la cual está habitualmente catalizada por una enzima llamada Taq polimerasa. Dicha enzima se aísla de una bacteria termófila, denominada Thermus Aquáticus, que es capaz de crecer a temperaturas elevadas (79 - 85 º C). A esta temperatura dicha enzima es capaz de mantener una media de extensión de más de 60 nucleótidos por segundo en regiones ricas en uniones G-C. La temperatura optima a la que actúa la Taq polimerasa permite el uso de elevadas temperaturas para la unión de los primers y para la extensión, de esta manera se aumenta el nivel de exigencia de la reacción y se reduce la extensión de los primers unidos inespecíficamente al ADN.
La reacción se lleva a cabo en una serie de ciclos cada uno de los cuales incluye tres fases o pasos:
DESNATURALIZACIÓN: Para que comience la reacción es necesario que el ADN molde se encuentre en forma de cadena sencilla. Esto se consigue aplicando temperaturas de 90 a 95ºC que producen la rotura de los puentes de hidrógeno intercatenarios y por lo tanto la separación de ambas cadenas. Para conseguir la completa separación de las hebras de toda la muestra esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse rápidamente, evitando así una eficiente hibridación de los primers y una posterior extensión.
HIBRIDACIÓN: Esta fase se denomina también fase de “annealing” o de emparejamiento. Una vez que el ADN está desnaturalizado se disminuye la temperatura hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la unión de los primers a las secuencias flanqueantes del fragmento que se va a amplificar. La temperatura de fusión o annealing (Tm, “melting temperature”) depende de varios factores y es relativamente específica para cada primer. La longitud de los primers y la secuencia son críticas en la designación de los parámetros de una amplificación, una fórmula simple para calcular la Tm es la siguiente:
Tm = 4(G+C) + 2 (A+T).
No obstante, cada primer exige una serie de estudios experimentales para determinar su temperatura de annealing específica ya que si la temperatura es muy baja la unión se hará de forma inespecífica y si es muy alta no se producirá una unión completa.
EXTENSIÓN: Durante este paso la Taq polimerasa incorpora nucleótidos en el extremo 3' del primer utilizando como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se lleva a cabo este paso suele ser de 72ºC ya que es la temperatura a la que la Taq polimerasa alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pb, y 40 segundos para fragmentos por encima de 1.2Kb.
Un factor importante en el transcurso de las diferentes fases es el tiempo de rampa. Este se define como el tiempo invertido en pasar de una temperatura a otra y depende del diseño y de las características del aparato donde se realiza automáticamente este proceso, el termociclador. En las nuevas generaciones de termocicladores este factor se ha ido optimizando para hacerlo mínimo.
Los buffer de PCR que se utilizan normalmente contienen KCl, Tris y MgCl2. El MgCl2 es el componente que más influye en la especificidad y rendimiento de la reacción ya que los iones Mg2+ son necesarios para la actividad de la Taq polimerasa, es decir, actúan como cofactores de la polimerasa.
La concentración óptima de MgCl2 está en torno a 1.5 mM si se emplean concentraciones de 200 mM de cada uno de los dNTPs. No obstante, a veces es necesario probar con diferentes cantidades de Mg ya que un exceso del mismo origina una acumulación de productos inespecíficos y una cantidad insuficiente hace que disminuya el rendimiento de la amplificación.
A la hora de elegir unos primers para amplificar un determinado fragmento de ADN hay una serie de reglas a seguir:
|
La longitud de cada uno de los primers debe estar comprendida entre 18 y 24 bases ya que se ha comprobado que primers de mayor longitud (30-35 bases) no aumentan el rendimiento y los primers cortos carecen de suficiente especificidad. | |
|
Ambos primers deben tener una Tm similar (como mucho la diferencia entre ambas temperatura debe ser de 5ºC). | |
|
La relación bases púricas: bases pirimidínicas debe ser 1:1 (o como mucho 40-60%). | |
|
La secuencia de los primers debe comenzar y terminar con 1-2 bases púricas. | |
|
Para evitar la formación de dímeros de primers es necesario comprobar que los primers no contengan secuencias complementarias entre sí. |
Los dímeros de primers consisten en fragmentos de doble cadena cuya longitud es muy próxima a la de la suma de los primers y se producen cuando un primer es extendido a continuación del otro. El mecanismo exacto por el que se forman estos dímeros no está completamente determinado. La observación de que primers con los extremos 3' complementarios favorecen su formación sugiere que el paso inicial se debe a interacciones transitorias que aproximan los extremos complementarios. Algunas polimerasas, incluida la Taq, han mostrado una débil actividad polimerizadora no dirigida por un ADN patrón, la cual puede unir nucleótidos adicionales al doble extremo apareado. Si esta actividad puede producirse sobre una hebra sencilla de oligonucleótidos, resultaría una buena oportunidad para que la extensión formara un corto solapamiento en el extremo 3' con el otro primer, suficiente para promover la formación del dímero.
Las concentraciones de dNTPs que suelen usarse están en torno a 200 µM para cada uno de ellos. En un volumen de reacción de 25 µl con esta concentración de dNTPs se sintetizarían entre 6-6.5 µg de ADN. La concentración de dNTPs y de MgCl2 va relacionadas ya que el Mg se une a los dNTPs con lo que concentraciones elevadas de dNTPs inhibirían la reacción al no tener la Taq polimerasa suficiente Mg como para incorporar dNTPs. Para una concentración de 200 µM de cada dNTP se suele añadir MgCl2 a una concentración de 1.5 mM.
Las cantidades óptimas de Taq polimerasa necesarias para la síntesis de ADN están alrededor de 2 unidades en 25 µl de volumen final de reacción. La actividad de este enzima se ve influenciada por la concentración de dNTPs, de Mg2+ y de algunos iones monovalentes de manera que concentraciones elevadas de los mismos inhiben dicha actividad.
Por otro lado, pequeñas concentraciones de KCl estimulan la actividad sintética de la Taq en un 50-60% con un máximo aparente cuando su concentración es de 50 mM. Existen algunos datos relacionados con la influencia de ciertos reactivos que se emplean antes de la amplificación y que alteran la actividad de la Taq. Por ejemplo concentraciones 1M de urea estimulan la actividad, el SDS a bajas concentraciones que la inhibe al igual que concentraciones mayores del 10% de etanol.
Es el ADN del cual queremos copiar un determinado fragmento, es, por tanto, el ADN que la Taq polimerasa utiliza como molde para la síntesis de nuevas cadenas polinucleotídicas. La cantidad de ADN necesaria para la PCR depende de varios factores:
Del marcador que se va a amplificar: hay marcadores cuyos primers son más específicos o bien cuyas condiciones de amplificación están mejor optimizadas que las de otros. Por esta razón puede darse el caso de que cierta cantidad de ADN (sobre todo cuando jugamos con cantidades mínimas) amplifique para unos marcadores pero no para otros. Por ello, cuando en un laboratorio se va a utilizar un nuevo marcador es necesario hacer un estudio de validación en él que se incluye un estudio de sensibilidad. De dicho estudio de sensibilidad puede sacarse como conclusión cuál es la mínima cantidad de ADN que amplifica en condiciones estándar. De manera general, para casi todos los STR utilizados en Genética Forense la cantidad óptima de ADN que asegura un rendimiento adecuado está en torno a los 5 ng.
Calidad del ADN: Cuando se trabaja con ADN cuya calidad es óptima no suele haber problemas en la amplificación y cantidades del mismo por encima e incluso por debajo de los 5 ng rinden buenos resultados. El problema aparece cuando la calidad del ADN obtenido no es la idónea, bien porque esté degradado o bien porque dicho ADN vaya ligado a una serie de contaminantes que pueden inhibir la actividad de la polimerasa. Si el ADN está degradado por la acción de enzimas de restricción el que obtengamos o no resultado en la amplificación va a depender de que el fragmento a amplificar haya sido dañado o no. En el caso en el que tengamos ADN sin degradar pero unido a una serie de contaminantes habría que intentar diluir al máximo la muestra para disminuir dichos contaminantes, pero siempre dentro de un rango de ADN que no esté por debajo del límite de sensibilidad de la PCR. El problema de las cantidades mínimas de ADN y la presencia de contaminantes o inhibidores de la Taq es un hecho habitual en Criminalística y requiere un estudio pormenorizado de la muestra antes de la amplificación.
Son elementos que mejoran el rendimiento y la especificidad de la PCR. Aunque algunos autores han recomendado el uso del DMSO y del glicerol, el adyuvante más extendido y utilizado es el BSA. A concentraciones por encima de 0.8 µg/µl el BSA incrementa la eficiencia de la PCR ya actúa como una proteína captadora de iones que pueden ser inhibidores de la Taq polimerasa.
La PCR ofrece una serie de ventajas, frente al uso de las técnicas de análisis genético utilizadas con anterioridad, como son:
|
Su capacidad para obtener resultados en casos en los que la cantidad de ADN es mínima o en casos en los que el ADN esté parcialmente degradado. | |
|
Genera en un espacio corto de tiempo un elevado número de copias de la secuencia de ADN que es objeto de estudio, lo cual permite utilizar técnicas de visualización más sencillas y rápidas que el uso de sondas marcadas radioactivamente. | |
|
Permite la determinación y agrupación alélica en clases discretas, lo que facilita la elaboración de bases de datos al ser la estandarización inmediata y posibilitar la aplicación de métodos bioestadísticos y programas elaborados. |
El uso de marcadores microsatélites de pequeño peso molecular aumenta las probabilidades de obtener resultados positivos de amplificación cuando el ADN se encuentra degradado ya que puede ser que dichos fragmentos no hayan sido digeridos. Esta ventaja es de gran importancia en Criminalística ya que normalmente los indicios biológicos encontrados han estado sometidos a diversos factores (calor y humedad) que favorecen el crecimiento bacteriano.
Sin embargo, una de las grandes ventajas de la PCR que es su elevada sensibilidad puede, en ocasiones, convertirse en un gran problema ya que se podría coamplificar un ADN extraño o ajeno al que nos interesa. No obstante, en los laboratorios de Genética Forense las medidas de precaución que se toman para evitar problemas de contaminación por manipulación son extremas.
Una vez amplificado el ADN, los fragmentos resultantes son separados en función de su tamaño por medio de un proceso de electroforesis. Actualmente se utilizan dos tipos de electroforesis en los laboratorios de Genética Forense:
|
Electroforesis en geles verticales de poliacrilamida | |
|
Electroforesis capilar |
La electroforesis capilar es una técnica relativamente novedosa en el campo de la Genética Forense pero que está poco a poco sustituyendo a los sistemas de electroforesis vertical. En este caso el proceso electroforético es llevado a cabo en un capilar de silica de unas 50 mm de diámetro, lo cual hace que la cantidad de calor generado sea menor y que puedan aplicarse voltajes mayores. Para que puedan ser analizados por electroforesis capilar los primers o los dideoxinucleótidos (en el caso de la secuenciación) deben ser marcados fluorescentemente con unas moléculas denominadas fluorocromos que emiten fluorescencia a una determinada longitud de onda cuando son excitados por láser. El equipo en el que se realiza el proceso lleva acoplado un ordenador encargado de traducir los datos de emisiones fluorescentes en secuencias o fragmentos con sus correspondientes alelos asignados. La electroforesis capilar presenta una serie de ventajas frente a los sistemas de electroforesis vertical como son:
|
Rapidez: ya que permite el análisis simultáneo de varios loci aunque éstos posean alelos con tamaños solapantes. | |
|
Sensibilidad: hace posible detectar cantidades muy pequeñas de ADN amplificado. | |
|
Los resultados se obtienen de manera informatizada, lo que evita problemas de interpretación de los resultados y facilita el análisis de los mismos a través de programas informáticos. |
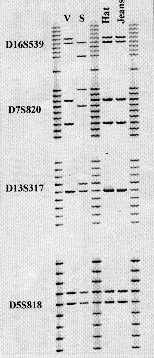 |
Ejemplo de un caso de criminalística resuelto utilizando electroforesis en gel vertical de acrilamida. Si se observan los patrones bandas podrá comprobarse como los perfiles obtenidos en las manchas de sangre encontradas en el sombrero (Hat) y pantalón (Jeans) del sospechoso (S) coinciden con el perfil genético de la víctima (V) |
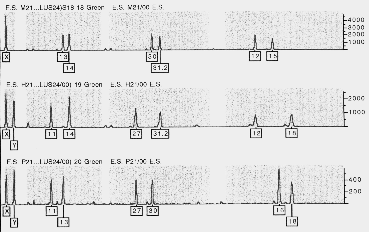 |
| Caso de estudio biológico de la paternidad realizado con electroforesis capilar. Cada pico corresponde a un alelo, el hijo (H) debe poseer un alelo de la madre (M) y otro del padre (P) para que la paternidad sea compatible |
![]()
Las técnicas de análisis genético se encuentran hoy en día en continuo desarrollo y evolución. La necesidad de técnicas que permitan el aislamiento y análisis de los casi cien mil genes que componen el genoma humano justifica la existencia de líneas de investigación destinadas al descubrimiento de nuevos métodos que permitan monitorizar elevados volúmenes de información genética en paralelo y que reduzcan tanto el tiempo empleado como el coste por análisis.
Desde el análisis de los primeros polimorfismos de ADN con fines identificativos, la Genética Forense ha sufrido una gran evolución. Los expertos en la materia han sido testigos de cómo el descubrimiento de la PCR revolucionó las técnicas de identificación genética. Es probable que la próxima revolución la constituyan los llamados biochips o microarrays.
Los biochips surgen como consecuencia de una combinación entre técnicas microelectrónicas empleadas para la fabricación de microprocesadores informáticos y materiales biológicos. En general puede decirse que la principal característica de los chips es su capacidad para generar información en muy poco espacio, ya que posibilitan el procesamiento de multitud de ensayos simultáneamente. Esta característica es la que hace que los biochips sean probablemente la tecnología del futuro en el campo de las investigaciones biomédicas.
La fabricación de los biochips es similar a la de los chips informáticos: por medio de la técnica denominada fotolitografía se depositan circuitos microscópicos sobre láminas de silicio. En el caso concreto de los biochips, estas láminas son de vidrio y lo que se deposita en dichas láminas son cadenas de ADN. Las láminas de vidrio presentan una serie de ventajas como son:
|
Posibilidad de unir las cadenas de ADN a la superficie del cristal, convenientemente tratada, mediante enlaces covalente. | |
|
Su capacidad para aguantar altas temperaturas y lavados de elevada fuerza iónica. | |
|
Al ser un material no poroso el volumen de hibridación puede reducirse al mínimo. | |
|
Su baja fluorescencia evita ruidos de fondo. |
Hay compañías comerciales que han desarrollado otras estrategias para la fabricación de biochips. No obstante, los más usados actualmente y con mayor número de aplicaciones son los basados en técnicas fotolitográficas.
Estos chips, como se dijo anteriormente, consisten en una pequeña lámina de vidrio que posee unos grupos reactivos, a los que posteriormente se unirán los nucleótidos, protegidos mediante una película realizada con un agente químico fotodegradable. Mediante una máscara que se sitúan sobre el chip, se logra enfocar un haz de luz hacia unas posiciones y regiones determinadas, degradando al agente químico protector en dichas zonas y dejándolo intacto en las zonas protegidas por la máscara. A continuación se añade al chip un medio que contiene uno de los cuatro nucleótidos que se unirá, mediante enlace covalente, al cristal con los grupos reactivos que hayan quedado desprotegidos. Cada grupo añadido lleva una molécula receptora fotodegradable.
Todo este proceso se va repitiendo con los diferentes nucleótidos y máscaras hasta generar unos oligonucleótidos con las secuencias que nos interesen. El siguiente esquema muestra el todo el proceso explicado anteriormente.
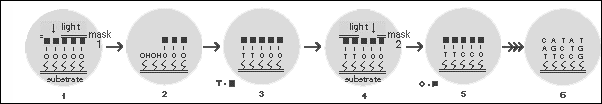
Cada “casilla” del chip posee una cadena de un oligonucleótido de manera que solamente aquel fragmento de ADN que hibride perfectamente con ella permanecerá unido tras los diversos lavados.
Previamente a la hibridación, el ADN de la muestra a estudiar debe haber sido amplificado y marcado fluorescentemente en uno de sus extremos. Una vez marcado se incuba en el recipiente que contiene al chip tras lo cual se lava varias veces para eliminar los fragmentos que no hayan hibridado y se introduce el chip en un escáner en el que se detectan los patrones de hibridación. Esta detección se realiza en base a la fluorescencia emitida por los fluorocromos de la muestra cuando son excitados por luz y en aquellos pocillos en los que la unión haya sido completa la fluorescencia será mayor que los que contengan alguna base desapareada.
Un ordenador conectado al escáner es el que identifica las secuencias sonda por su posición el chip.

![]()
A pesar de ser una tecnología muy reciente y que, por lo tanto, está aún en vías de experimentación, actualmente los biochips están siendo aplicados en:
|
Monitorización de expresión génica: permite determinar cual es el patrón de expresión génica y cuantificar el nivel de expresión de manera simultánea para un elevado número de genes. Esto permite realizar estudios comparativos de activación de determinados genes en tejidos sanos y enfermos y determinar así la función de los mismos. | |
|
Detección de mutaciones y polimorfismos: Permite el estudio de todos los posibles polimorfismos y la detección de mutaciones en genes complejos. | |
|
Secuenciación: Mientras que se han diseñando algunos biochips para secuenciación de fragmentos cortos de ADN, no existe aún en el mercado ningún biochip que permita secuenciar de novo secuencias largas de ADN. | |
|
Diagnóstico clínico y detección de microorganismos: Posibilitan la identificación rápida empleando unos marcadores genéticos de los patógenos. | |
|
Screening y toxicología de fármacos: el empleo de los biochips permite el analizar los cambios de expresión génica que se dan durante la administración de un fármaco de forma rápida, así como la localización de nuevas posibles dianas terapéuticas y los efectos toxicológicos asociados. | |
|
Seguimiento de terapia: los biochips permiten valorar rasgos genéticos que pueden tener incidencia en la respuesta a una terapia. | |
|
Medicina preventiva: El conocimiento y posible diagnóstico de ciertos caracteres genéticos asociados a determinadas patologías permite una prevención de las mismas antes de que aparezcan los síntomas. |
![]()
Copyright © 2000 by Carmen Entrala. Dra. en biología.
![]()
Otros documentos del Curso de Doctorado: