|
Ab initio (MP2/6-311+G*) and density functional theory (B3LYP/6-311+G*) calculations
have been performed to determine the bonding nature of N, P and As (pnicogen) with B or C in ylides
and their boron analogs. The compounds studied refer to the formulas R3XCR'2
and R3XBR' (X=N, P,
As; R=H, F; and R'=H, SiH3). The computed electron density has been analyzed by means of the Atoms
in Molecules (AIM) theory and the Electron Localization Function (ELF) methods. In addition, the
rotational barriers were calculate for the X-C and X-B bonds at the CASMP2/6-311+G* level to
elucidate the multiple bonding character for these bonds. The geometrical and electronic results
indicated that the N ylides differ remarkably from the remaining pnicogen (P, As) ylides, the former
yielding clear single bonds, while the latter showed stronger multiple bonds. Moreover, the fluorine
substituents strengthened the X-C and X-B bonds, reducing the bond distance, increasing the electron
density and augmenting the planarity at the C and B atoms. However, the SiH3 groups affected only the
planarity at the C atoms for the organic ylides. This indicates how the electronegativity of the different
substituents influence the central X-C and X-B bonds: if the substituent pulls charge from the bond in
the direction towards the pnicogen, the bond is reinforced and it is more likely to present double-bond
characteristics. This is not accomplished by substituents pushing charge in the aforementioned direction.
Differences between the organic ylides and their boron derivatives have been found. Boron analoges
presented a remarkable asymmetric X-B bonds, with a rotation barrier of ca. 30 kcal/mol, caused by a
strong repulsion between the lone pairs of the XH3 unit and that of boron.
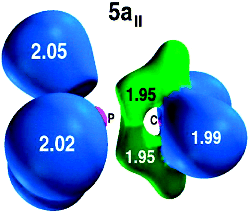 ELF representation at 0.7 for rotamer 5a|, at the B3LYP/6-311+G*//B3LYP/6-311+G* level.
|