Texto tomado fundamentalmente de
Reed, Stephen J.B., 2006. Electron Microprobe Analysis and
Scanning Electron Microscopy in Geology, 2e édition.
Cambridge University Press, Cambridge, 206 p.
Xavier
Llovet, Aurélien Moy, Philippe T. Pinard, John H. Fournelle (2021).
Electron probe microanalysis: A review of recent developments and
applications in materials science and engineering. Progress in Materials
Science 116, 100673.
Cuantificación de la señal
Los RX característicos de un elemento dado se dirigen a un detector de
rayos X (dentro junto al espectrómetro y de la configuración geométrica
del
círculo de Rowland
) donde se produce una señal eléctrica (generalmente, en un
contador
de flujo continuo de gas). Esta señal es
proporcional al número de rayos X, que a su vez es proporcional a la
concentración del elemento "i" en la muestra. La señal eléctrica se
amplifica y se compara con la señal producida en las mismas condiciones
por un estándar de composición conocida. La concentración final del elemento "i" en la
muestra se calcula basándose en la proporcionalidad entre la intensidad
de los rayos X en la muestra desconocida y la intensidad de los rayos X
producida por el estándar conocido, siguiendo un procedimiento de
corrección para minimizar el efecto "matriz" en la medición.
El espectro de rayos X se utiliza para análisis cuantitativos midiendo la
intensidad de los picos característicos de rayos X correspondientes a diferentes
elementos de la muestra siguiendo los siguientes pasos:
-
Identificación y medición de la intensidad de pico: El primer paso
consiste en identificar los picos característicos en el espectro,
que corresponden a elementos específicos. El software de las
máquinas permite identificar automáticamente los picos y, en
general, deconvolucionar con precisión los picos superpuestos
mediante métodos matemáticos. Se calcula
la intensidad (cuentas por segundo por nanoamperio) de los picos
característicos de un material dado mediante los detectores de RX
característicos difractados. La intensidad está directamente
relacionada con la abundancia de los elementos correspondientes en
la muestra.
-
Sustracción de fondo: El fondo continuo,
correspondiente a la radiación de frenado (Bremsstrahlung), se
sustrae del espectro para medir con precisión las intensidades de
los picos característicos.
-
Estandarización y calibración: El análisis
cuantitativo requiere calibración con estándares de composición
conocida para comparar sus picos con los de la muestra. Estos
estándares ayudan a controlar la calidad de los cristales
analizadores y detectores (en EDS, determinar la eficiencia del
detector).
-
Corrección de efectos de matriz: Se aplican
correcciones para tener en cuenta los efectos de matriz ZAF, que
incluyen efectos de número atómico (Z), absorción (A) y
fluorescencia (F) que alteran la intensidad de los rayos X. Estas
correcciones garantizan que las intensidades medidas reflejen con
precisión las concentraciones reales de los elementos.
-
Cálculo cuantitativo y criterios de calidad: Utilizando las
intensidades de pico corregidas y los datos de calibración, se
calcula la concentración de cada elemento en la muestra. Esto puede
realizarse mediante diversos algoritmos cuantitativos y el software
incluido con el sistema.
1a.- Identificación y medición de picos
Lo ideal sería recopilar la señal total de cada pico
integrando todos los canales (longitudes de onda) bajo él. Para WDS,
esto implicaría un barrido angular del espectrómetro, lo que resulta
demasiado lento para la adquisición de datos cuantitativos o el
mapeo de rayos X. Por lo tanto, se mide un único canal centrado en
el pico y se asume que es representativo del área total bajo el pico
(en EDS se mide el área integrada del pico).

Espectros de RX difractados por TAP y PET en material cerámico con Si,
Al, Fe, Sn, Zr (Jeol).
La intensidad de los rayos X producidos por el elemento “i” en la
muestra se puede expresar como el número de rayos X contados por segundo
(cuentas/seg = cps) por el detector. Se conoce la longitud de onda de
los rayos X característicos de todos los elementos, así como el senoӨ (Ө
es el ángulo de difracción para un plano de espaciado dado, según la ley de
Bragg).
Las sondas Cameca controlan el movimiento del espectrómetro y la
indexación de rayos X basándose en valores de senoӨ. Las sondas JEOL
funcionan con el valor “L”, donde L es la longitud que conecta la
muestra con el cristal analizador (o sea, la longitud recorrida por el
rayo X difractado desde la muestra hasta el cristal analizador), L = 2R·senoӨ, siendo R el radio del círculo de Rowland (160 mm para las sondas
Cameca y de 140 mm para las sondas JEOL, excepto para los espectrómetros
“H” en las sondas JEOL, que tienen R=100 mm). Al ajustar la posición del
cristal analizador a un valor único de “sinӨ” o “L” para un elemento
“i”, el cristal analizador difractará únicamente los rayos X
de longitud de onda característica provenientes del elemento “i” (esto sucede porque los rayos X que tienen
una longitud de onda similar al espaciamiento d de la red cristalina del
cristal analizador serán refractados). Este es el principio que hizo posible
la invención de la microsonda electrónica por el estudiante de doctorado
francés Raymond Castaing en 1952. Los rayos X producidos por el elemento
“i” pueden así contarse durante un período de tiempo específico (“tiempo
de contaje”). Dado que el cristal de un espectrómetro WDS solo puede
registrar conteos de rayos X para una longitud de onda a la vez, el
análisis de múltiples elementos en un solo espectrómetro debe realizarse
secuencialmente,
comenzando por
los elementos de Z menor como el Na que puede verse afectados por
difusión fuera del volumen irradiado por fuerzas electrostáticas
acumuladas. Además, dado que ningún tipo de cristal puede
satisfacer la Ley de Bragg para todo el rango de longitudes de onda de
rayos X emitidos por todos los elementos de la tabla periódica, las
microsondas electrónicas están equipadas con varios espectrómetros
(4-5), con la capacidad integrada de intercambiar automáticamente
diferentes cristales analizadores dentro de un mismo espectrómetro
(generalmente, hasta diez cristales difractores en total para registrar
todos los elementos deseados con Z ≥ 4). Todo ello ralentiza la adquisición de datos
para el análisis (generalmente, unos 5-10 minutos por análisis puntual).
1b.-
Interferencias espectrales y superposiciones
En ocasiones, las intensidades de los picos se ven potenciadas por la
superposición de un pico vecino.
Las interferencias espectrales son un problema para EDS- y WDS-EPMA (y
SEM-EDS), ya que difuminan los picos elementales de rayos X de interés,
requiriéndose una corrección de superposición. En cualquier caso, las
interferencias pueden resolverse mejor en WDS que en EDS, dada la mayor
resolución de energía
La cantidad de superposición puede expresarse como una fracción de la
intensidad del pico principal del elemento superpuesto. En EDS esto
afecta a toda el área debajo de la curva del pico.
Con WDS, el aumento de la resolución espectral reduce muchas de estas
interferencias ya que trabaja con canales estrechos de longitud de onda.
Pero, a diferencia de EDS, el uso de la difracción de Bragg para WDS genera
problemas de "n mayor que uno", por lo que los
espectros de WDS contendrán cierta cantidad de estos picos de alto orden
de difracción (n > 1). A veces, pueden coincidir con un pico objetivo o
muy cerca de él, con una longitud de onda más baja o más alta, y
potencialmente ser convertirse en interferencia para la medición del
fondo.

Distintas respuestas de interferencia (rojo) en función de la posición
de los picos de elemento medido (verde) y del elemento interferente
(azul). Las respuestas se deben a la suma de intensidades de longitud de
onda de los elementos medido e interferente. Una vez superada cierta
separación de picos (Dl3), no hay
interferencia en el pico del elemento analizado.
Se utilizan varios métodos para corregir las interferencias espectrales. El método más sencillo consiste en medir la intensidad de los
rayos X en la posición (longitud de onda) de la línea superpuesta (p.ej.,
V ka) en una muestra de
referencia (patrón, por ejemplo, Ti puro) que contiene el elemento que se superpone
(p.ej., Ti), pero no
el superpuesto (p.ej., V). Esta intensidad se resta de la
intensidad medida en el elemento desconocido en una proporción
constante, que debe calcularse del análisis del patrón. Por ejemplo, la
presencia de Ti oscurecerá la concentración de V en una muestra
que contenga ambos elementos ya que V Ka se
ve interferida por Ti Kb. La interferencia V
Ka - Ti Kb se resuelve midiendo las intensidades de Ti Ka
y Ti Kb
en un patrón (Ti puro) y en el material desconocido que contiene V de
forma que, dado que la relación de intensidades de Ti Kb
y Ti Kb
es la misma en ambas matrices:
(I TiKb/I TiKa)PATRON
= (I TiKba/I TiKa)DESCONOCIDO
= constante
la intensidad real V Ka en
el material desconocido es:
I VKaREAL =
I VKaTiKbMEDIDA
- I TiKbDESCONOCIDO
I VKaREAL =
I VKaTiKbMEDIDA
- [I TiKaDESCONOCIDO · (TiKb/TiKa)PATRON]
siendo (TiKb/TiKa)PATRON
el factor de intensidades e VKaTiKbMEDIDA
la intensidad medida en la línea interferida, por oposición a la real.
Así, en general, los factores de intensidades pueden determinarse a
partir de patrones de elementos puros, y la intensidad real de la línea
interferida se encuentra restando de la intensidad medida lo calculado
utilizando una intensidad de referencia del elemento que interfiere y el
factor de intensidades del patrón:
IREAL = IMEDIDA - [IDE
REFERENCIA DESCONOCIDO · Factor de IntensidadesPATRON]
Lo anterior aplica a espectros EDS y WDS, si bien, como se muestra más
abajo, en WDS puede que no hiciera falta la corrección por
interferencias ya que la resolución de energía de éste último método es
mucho mayor, pudiendo resolverse las medidas de interés en el espectro
WDS de forma satisfactoria.

Espectro EDS de una aleación Ti-6Al-4V que muestra la superposición
observada entre las líneas Ti Kb (4.932 keV)
y V Ka (4.952 keV) (de
Oxford Instruments).
En EDS es imposible resolver la interferencia.

Barrido de longitud de onda WDS (expresado en energía) para la aleación
Ti-6Al-4V mostrando picos distintivos de Ti Kb
y V Ka (morado). El espectro EDS se muestra
al fondo (amarillo) para fines comparativos. Como la resolución de
energía de WDS es 10 veces mejor que la de EDS, los picos superpuestos
de Ti y V se pueden diferenciar y las concentraciones de estos elementos
se pueden determinar con mayor precisión (de
Oxford Instruments).
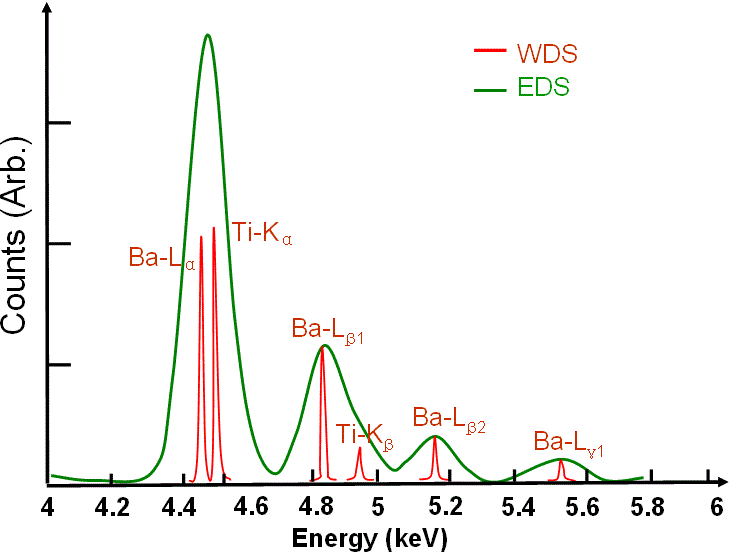
Interferencia Ti-Ba Espectro EDS de BaTiO3 con solapamiento
de picos EDS que se resuelven en WDS dada su mayor resolución de energía
(5-10 eV) (de Liao
2006).

Sección del espectro de rayos X de galena en la región de energía de 2.2
a 2.6 keV que muestra la alta resolución espectral de WDS en comparación
con EDS (de
Bruker)

Espectro WDS simulado para monacita que muestra un alto número de
solapamientos de líneas de pico entre los elementos de las tierras
raras. Se ha seleccionado la posición de pico del praseodimio (Pr Lb,
menos intenso que Pr La) para evitar el
solapamiento de los picos La Lβ y Pr Lα (de
Oxford Instruments).
Otros métodos más precisos incluyen la deconvolución espectral,
utilizando un espectro adquirido en una muestra de referencia conocida
que no contiene el elemento superpuesto. Por ejemplo, en un espectro U-Pu,
la línea de rayos X Pu Mβ se superpone con la línea U M3N4 de baja
intensidad (tenga en cuenta que la línea de rayos X Pu Mα no puede
utilizarse para el análisis debido a la fuerte interferencia con la
línea de rayos X U Mβ). La deconvolución, utilizando el espectro medido
a partir de un estándar de UO₂ y escalado adecuadamente, permite una
medición precisa de la intensidad Pu Mβ.
Las reflexiones Bragg de orden superior (n > 1) no deben ignorarse ya
que potencialmente constituyen una fuente de interferencia espectral.
Pueden generar problemas críticos para el trabajo con elementos traza.
En general, es posible minimizar la influencia de los picos de
difracción de orden >1 hasta en un ~90 % mediante el ajuste adecuado de
una ventana de energía en los contadores de RX (PHA, Pulse Height Analyzer), si bien debe
evitarse la supresión involuntaria de la altura del pulso de picos n =
1.

Armónicos de orden de difracción 1 y 2 en cobre difractado por el cristal LiF (2d
= 0.4026 nm), expresado en cuentas por segundo en función del ángulo de difracción).
Nótese que tanto para n = 1 como n = 2, la longitud de onda y la energía de Kα = 1.54
A = 8.06 keV y
de Kβ = 1.39 A = 8.92 keV (de
physicsopenlab.org).
|
|
n |
2θ (°) |
θ (°) |
senθ |
2d (nm) |
λ (A) |
E (keV) |
|
Cu Ka
(LIF) |
1 |
44.83 |
22.415 |
0.3813 |
0.4026 |
1.535 |
8.08 |
|
Cu Ka
(LIF) |
2 |
100 |
50 |
0.7660 |
0.4026 |
1.542 |
8.04 |
|
Cu Kb
(LIF) |
1 |
40.17 |
20.085 |
0.3434 |
0.4026 |
1.383 |
8.96 |
|
Cu Kb
(LIF) |
2 |
87.67 |
43.835 |
0.6926 |
0.4026 |
1.394 |
8.89 |
Las interferencias de pico deben corregirse, y el método más preciso
requiere iterarlas dentro de la corrección de matriz, en lugar de
restarlas como una corrección posterior a la corrección de matriz ZAF (o
PAP).
1c.-
Elementos traza
Los elementos traza pueden determinarse en roca-total mediante técnicas
de análisis químico como XRF, ICP-MS, etc. Sin embargo, para determinar
su distribución en las diferentes fases del material, se requiere una
técnica de análisis de microhaz como EPMA o LA-ICP-MS.
La emisión de Bremsstrahlung o de frenado se produce cuando el los electrones
incidente desaceleran al interactuar con el campo electrostico que rodea
los
átomos de objetivo, perdiendo energía en forma de rayos X emitidos y
creando una señal continua de fondo de rayos X (hasta el
límite de Duane-Hunt, que coincide con
la energía E0 de los electrones incidentes). Esta radiación
de frenado es el factor limitante para la detección de elementos traza
ya que el fondo generado puede ocultar los picos característicos de
estos elementos.
La cantidad de radiación de fondo aumenta con la
densidad de la muestra, lo que significa que los elementos ligeros en
una matriz pesada pueden ser difíciles de detectar. La
forma de la radiación de fondo, descendiendo de intensidad hacia altas
energías, hace que los elementos en traza pesados puedan detectarse con
mayor facilidad que los ligeros.

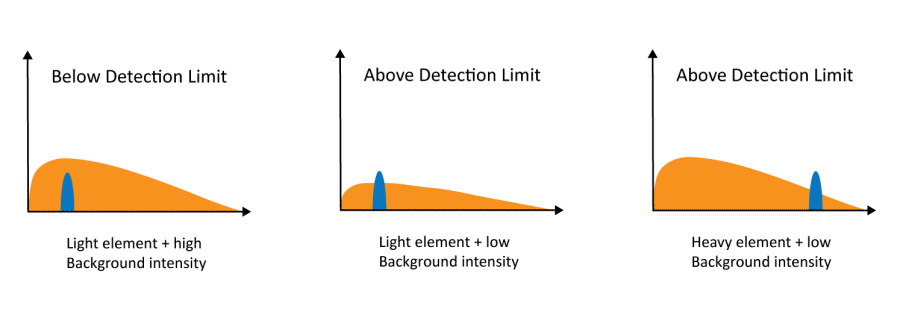
El espectro de rayos X está compuesto por picos de rayos X
característicos y un fondo continuo conocido como Bremsstrahlung
(radiación de frenado). La intensidad del fondo está determinada por la
densidad de la muestra. Por ello, puede ser difícil detectar trazas de
elementos ligeros en una muestra con una matriz de elementos pesados. De
Nanoscience Instruments.
El parámetro más importante para el análisis de trazas es el límite
de detección. Se trata de cuanto de pequeña puede ser una variación
estadística normal del fondo para poder identificarla con un pequeño
pico característico. Por tanto, el análisis de trazas mediante EPMA necesita una
resolución espacial muy
alta y una alta sensibilidad. Esto presenta numerosos desafíos, si bien
la precisión no es el mayor obstáculo, porque los instrumentos modernos
ofrecen una buena eficiencia de recolección de RX y una corriente de
sonda alta y estable, lo que permite adquisiciones prolongadas. La
resolución espacial del análisis puede extenderse muy por debajo de 1 μm
en análisis de bajo voltaje (por debajo de 15 kV) de fases de alto
número atómico promedio. El uso de alta corriente de sonda y/o un tiempo
prolongado de exposición del haz para lograr análisis de alta
sensibilidad puede agravar los efectos de daño y carga, especialmente en
materiales aislantes como las rocas/minerales, lo que puede comprometer
la precisión. Un voltaje más alto mitiga parte de esto y aumenta las
tasas de conteo, pero las desventajas obvias son la menor resolución
espacial y las altas correcciones de la matriz.
El análisis de trazas mediante EPMA es fundamentalmente una
caracterización y medición del fondo, ya que pequeñas imprecisiones en
la determinación del fondo producen grandes errores en la intensidad
neta. La curvatura de fondo y las interferencias hacen que la
interpolación lineal simple de 2 puntos sea inadecuada (especialmente en
materiales complejos), lo que puede resultar en errores de decenas a
cientos de ppm.
De
M J Jercinovic, M L Williams, J Allaz y J J Donovan (2012). Trace analysis in EPMA.
La intensidad de los rayos X viene regulada por la sobretensión (U),
dado que la probabilidad de emisión de rayos X aumenta a medida que
aumenta la tensión de aceleración (voltaje). La relación pico-fondo P/B
de un material dado utilizando un cristal WDS dado (o un detector EDS
específico) puede aproximarse mediante:
P/B = 1/Z(U-1)n
donde U es la relación de sobretensión (E0/Ec) y n
es un exponente que normalmente se encuentra en el rango de 0.3 a 0.7.
De esta ecuación se deduce que la relación pico-fondo P/B aumenta al
aumentar la tensión de aceleración. Por lo tanto, al operar el
instrumento EPMA con una sobretensión alta, se puede superar el límite
de detección para una corriente de sonda y un tiempo de contaje
determinados será menor. Cabe destacar que los dos últimos factores
(corriente de sonda y tiempo de contaje) determinan la precisión de los
análisis, y es la reducción de la variabilidad analítica del fondo la
que permite medir con precisión concentraciones muy pequeñas. WDS tendrá
límites de detección más bajos que el EDS, debido a su mejor resolución
espectral y, por consiguiente, a una mayor relación pico-fondo.

Relaciones cualitativas Pico/Fondo (P/B) en función del número atómico y
el sobrevoltaje.
En general, usando energía estándar (15-30 keV), el análisis de
elementos en traza necesita de
-
alta corriente sonda (varios cientos de nA y hasta algo más de mil nA,
-
altos tiempos de contaje de pico
(centenas de segundos) y
-
deben agregarse los datos de varios
espectrómetros (más de un espectrómetro midiendo al mismo tiempo el
mismo elemento)
para alcanzar límites de detección de varias unidades a
decenas de ppm. Vea, p.ej.,
Dominguez-Carretero et al. (2025) Accurate electron probe microanalysis of key petrogenetic minor and trace elements in Cr-spinel, 10.1016/j.chemgeo.2024.122579.
Para evaluar la precisión de los análisis de trazas en EPMA, es habitual
comparar los resultados con los de otras técnicas microanalíticas, como
PIXE, μ-XRF, SIMS o LA-ICP-MS. Por ejemplo,
Sobolev et al. 2007
midieron sistemáticamente varios elementos traza en olivino de rocas
basáticas para investigar la contribución de fuentes corticales
recicladas en el manto. Estos autores analizaron más de 17000 granos de
olivino y reportaron límites de detección de 6 a 15 ppm y errores de 15
a 30 ppm (a nivel 2σ) para los elementos Ni, Ca, Mn, Cr, Co y Al. La
precisión de los resultados de EPMA se evaluó comparándolos con los
resultados de LA-ICP-MS. Un procedimiento similar ofrecen
Dominguez-Carretero et al. (2025)
para elementos traza en cromititas.

Precisiones analíticas (2s) para Ti, V, Ni y
Mn y (b) límites de detección (3s) para Sc,
V, Ti, Mn, Co, Ni, Zn y Ga, obtenidos sobre un patrón de referencia de
Cr-espinela en función de la corriente de sonda, a 25 keV y tiempos de
contaje de pico y fondo de 60 y 30 segundos, respectivamente.
Dominguez-Carretero et al. (2025).
1d.-
Tiempo muerto
Dado que la electrónica tarda un tiempo finito (del orden de
microsegundos) en procesar un evento (detectar un pulso generado por
rayos X), existe un período en el que está "ocupado" con una señal y
debe ignorar las demás. Por lo tanto, el detector es ciego a la llegada
de nuevos fotones durante este "tiempo muerto". Los tiempos
muertos típicos están en torno a unos pocos ms
(microsegundos).
Cuando la tasa de la
señal de rayos X es baja, es improbable que llegue un segundo fotón
durante el tiempo "ocupado". Cuando la tasa de la señal es alta, una
fracción significativa del tiempo real se dedica al procesamiento de
eventos y esta fracción de tiempo real se registra tiempo muerto. El
software corrige los conteos medidos para el tiempo muerto.
Todos los sistemas de detección de rayos X deben incluir en sus esquemas
de cuantificación una corrección del tiempo muerto; es decir, tener en
cuenta la incapacidad de la electrónica del detector para contabilizar
correctamente cada rayo X, debido a la fracción finita de tiempo en la
que el sistema está "ocupado" y no detecta rayos X. Los fabricantes de
instrumentos de sonda electrónica proporcionan una "constante de tiempo
muerto" para cada espectrómetro/detector suministrado. Sin embargo, para
una cuantificación precisa, algunos laboratorios de sonda electrónica
han determinado valores más precisos para sus instrumentos específicos y
han descubierto que los valores de tiempo muerto cambian con el
envejecimiento del instrumento y no son constantes para todas las
corrientes de haz/velocidades de conteo.
El tiempo muerto definido como el intervalo de tiempo (t) tras la
llegada de un pulso durante el cual el sistema no responde a pulsos
posteriores (normalmente unos pocos microsegundos), hace que la tasa
de conteo medida (n') sea menor que la tasa de conteo real
(n) en una cantidad que se vuelve significativa a tasas de conteo altas,
según la ecuación:
n' = n(1-n·t)
Las intensidades medidas pueden corregirse por tiempo
muerto aplicando esta ecuación. Por ejemplo, con un tiempo muerto típico
de 3 ms (30-6 s), la medición de
30000 cuentas/s supone corregir en 10% (3000 cuentas/segundo), o sea,
las cuentas reales serían 33000 cuentas/s.

Intensidades reales vs
medidas en función del tiempo muerto.
2.-
Modelado y sustracción del fondo
El análisis cuantitativo con microsonda se basa en la comparación entre
a) la intensidad medida (cps netas, Iiunk) de los
rayos X de cada elemento “i” presente en una muestra desconocida, y b)
la intensidad medida (cps netas, Iistd) de los
rayos X del mismo elemento “i” en un material estándar cuya
concentración se conoce. Pero la intensidad de pico medida incluye una
contribución del fondo, originada principalmente en el continuo
de rayos X. Por ello, la comparación debe realizarse entre las intensidades netas de los rayos X en el elemento desconocido y el
estándar durante el
“tiempo
de contaje”, donde la intensidad neta (Ii) es la diferencia
entre la intensidad de pico total (IPi) y el promedio de las intensidades de los
fondos inferior y superior (IBi), tanto en el
estándar como en la muestra desconocida:
Iiunk = IPiunk - IBiunk
Iistd = IPistd - IBistd
El fondo varía lentamente en función de la longitud de onda y puede
estimarse tomando el promedio de las mediciones a cada lado del pico
(fondo inferior y superior). La curvatura del fondo es insignificante
para la mayoría de los propósitos, pero puede ser significativa en la
región de la cola de un pico principal vecino. Si las mediciones de
fondo aumentan por la superposición de un pico vecino, la intensidad
aparente pico-fondo es demasiado baja e incluso puede ser negativa. Este
efecto puede evitarse midiendo el fondo solo en un lado, donde no hay
superposición pero debe tenerse en cuenta la pendiente. La cola
del pico adyacente se puede representar mediante polinomios de orden
variado.

Fondo determinado promediando las intensidades medidas en posiciones de
desplazamiento (fondo1 y fondo2): (a) fondo lineal: se obtiene el
resultado correcto para el fondo bajo el pico; (b) fondo curvo: se
obtiene un resultado incorrecto si se interpola linealmente (Reed,
2006).
El procedimiento estándar para obtener la intensidad neta de pico en una
medición WDS consiste en registrar la tasa de conteo en el canal
correspondiente al centro/máximo del pico y, habitualmente, aunque no
siempre, en dos posiciones de fondo a ambos lados del pico (se
permite usar solo un lado si las condiciones lo requieren y es posible
modelar el fondo correctamente). La intensidad de fondo en la intensidad
máxima del pico se obtiene mediante la interpolación de las mediciones
de fondo. De forma óptima, el software permite diferentes opciones de
modelo de fondo, desde lineal hasta curvo/exponencial y con pendiente,
que se resta de la tasa de conteo medida. Este procedimiento
proporciona, en general, resultados precisos gracias a la alta relación
pico-fondo de los espectros WDS.
La sustracción precisa del fondo es fundamental, en particular para las
concentraciones menores y traza. El continuo en el espectro WDS no es
necesariamente una línea recta de un extremo al otro, sino que puede
mostrar una curvatura de leve a significativa. Un modelado adecuado del
fondo es fundamental. Hay dos cuestiones:
(i) Puede haber interferencias
de primer orden o de orden superior en las posibles ubicaciones del
fondo, que deben evitarse. No existe una regla que indique que se
debe colocar una posición de fondo en el lado inferior y otra en el lado
superior del pico. Es perfectamente correcto tener dos fondos en un
lado del pico. La única regla es que el modelo de fondo bajo la
posición característica del pico debe ser coherente con la
forma/pendiente del fondo a medida que transita de un lado al otro del
espectro.
(ii) No todos los fondos son lineales, y muchos, especialmente
en el extremo inferior del espectro, son curvos.
Por lo tanto, es esencial que el software utilizado sea capaz de
proporcionar cierto grado de curvatura en el modelo de fondo.
Obviamente, si el fondo es curvo, no se puede asumir que es recto (se
determinaría un fondo incorrecto, demasiado alto, lo que resultaría en
una subestimación de la intensidad neta y, por lo tanto, de la
concentración).

Espectros de longitud de onda representativos alrededor de los picos de
interés obtenidos en uno de los granos de Cr-espinela adquiridos en las
condiciones indicadas en cada panel con indicación de las posiciones
donde se midió el fondo.
Domínguez Carretero et al.
(2025). Accurate electron probe microanalysis of key petrogenetic minor and trace elements in Cr-spinel.
Para un
rango limitado de mediciones, por ejemplo, elementos mayores Si, Ti, Al,
Fe, Mn, Mg, Ca, Na y K en silicatos comunes se
puede determinar fácilmente un conjunto rutinario de compensaciones del
fondo. Pero para materiales no comunes es fundamental determinar las
compensaciones óptimas de fondo caso por caso mediante la
adquisición de escaneos de longitudes de onda, aunque programas como
"Virtual WDS" y la "herramienta de desarrollo de métodos EPMA"
pueden simplificar la tarea.
3.-
Calibración y comparación con estándar (razón k)
Castaing sentó las bases del análisis cuantitativo en EPMA reconociendo
que con esta técnica era esencial medir la intensidad de los rayos X de
un material de referencia estándar utilizando exactamente
el mismo equipo y las mismas condiciones analíticas que para medir el
material desconocido, lo que llevó a definir la razón k.
Para cualquier elemento “i”, la razón entre la intensidad neta del
elemento “Iiunk” en la muestra desconocida y la
intensidad neta del elemento “Iistd” en el
estándar (Iiunk/Iistd) se
conoce como razón ki. Por lo tanto, la concentración
(% peso) del elemento “i” en la muestra desconocida (Ciunk)
se da con la aproximación conocida como Primera Aproximación de
Castaing:

Por ejemplo, si en las mismas condiciones obtenemos una intensidad neta
de 20,000 cuentas/seg para Fe en una muestra desconocida y 40,000
cuentas/seg para Fe en un estándar que contiene 50 % en peso de Fe, en
una primera aproximación, el contenido de Fe en la muestra desconocida
se calcula como:
(20,000/40,000) * 50 % en peso de Fe
= ½ * 50 = 25 % en peso de Fe.
Castaing, además de crear la primera sonda electrónica operativa,
desarrolló el procedimiento analítico utilizando metales elementales
puros como patrones y relacionando el contaje de rayos X de un
elemento en la aleación desconocida con el contaje de rayos X del
metal elemental puro. El uso de estos patrones de referencia en EPMA es
fundamental para la precisión de las mediciones de materiales
desconocidos. Diversas agencias han definido la terminología de los
patrones (p. ej., el
Instituto Nacional de Estándares y Tecnología de EE. UU., el
Comité Conjunto de
Guías en Metrología)
Para el microanálisis, un Material de Referencia se define como
un material suficientemente homogéneo (es decir, a escala
micrométrica) y estable bajo el haz de electrones. Su composición
química debe conocerse, preferiblemente mediante una técnica diferente
(p. ej., análisis químico húmedo, XRF, etc.). Un Material de
Referencia Certificado es un Material de Referencia (MR) que se
acompaña de un certificado que proporciona la composición química, la
incertidumbre asociada y una declaración de trazabilidad metrológica.
Algunas agencias, como el NIST, pueden proporcionar niveles de
certificación adicionales, por ejemplo, el Material de Referencia
Estándar del NIST.
Normalmente, en la ciencia de los materiales, se utilizan metales de
pureza definida en muchas aplicaciones; también se utilizan compuestos
binarios y ternarios (por ejemplo, semiconductores). Además, se utilizan
boruros, carburos, nitruros y óxidos. Para los casos en que se analizan
silicatos, carbonatos, fosfatos y otros materiales cristalinos, se
utilizan como estándares muchos minerales naturales y sintéticos. Diversos
vidrios de referencia también son útiles como estándares.
Existen diversas fuentes de estándares microanalíticos; por ejemplo,
varios proveedores de de microscopía electrónica o
institutos gubernamentales de estándares venden bloques montados. Estos
deben ir acompañados de la documentación correspondiente que indique la
composición y la fuente. También se pueden adquirir granos individuales,
montarlos y pulirlos adecuadamente.
El uso y cuidado adecuados de los estándares microanalíticos a veces no
se valora lo suficiente. En general, la coincidencia
de matrices de los estándares con las muestras desconocidas es esencial para el
análisis EPMA de compuestos que combinan elementos
de bajo y alto Z.
Vea también
apéndice en Llovet et al. (2021).
4a.-
Corrección de matriz ZAF
En la práctica, la primera aproximación de Castaing debe corregirse para
tener en cuenta los efectos de otros elementos presentes en la muestra
(y el estándar), que pueden afectar de forma diferencial la producción
de rayos X (es decir, efectos de la matriz). La corrección de los
efectos de la matriz más común se denomina corrección ZAF y fue
propuesta por primera vez por Castaing. Esto significa que la
concentración del elemento "i", calculada mediante la primera
aproximación, debe multiplicarse por el cociente ZAF, donde:
Z es un factor de corrección para
diferentes elementos con diferente número atómico Z presentes en la
muestra desconocida y el estándar.
A es un factor de corrección para la
absorción diferencial de rayos X.
F es un factor de corrección para la
generación diferencial de fluorescencia secundaria.
El punto de partida de un método
cuantitativo es establecer la relación entre la intensidad de rayos X y
la concentración de elementos. Esto puede lograrse con dos enfoques
diferentes:
(i) integrar cada contribución a
lo largo de la trayectoria del electrón y considerar la pérdida
de generación de rayos X debido a la retrodispersión de electrones,
y
(ii) integrar cada contribución a
lo largo de la profundidad de la muestra, siempre que se conozca
la distribución de la emisión de rayos X en profundidad.
El primer enfoque es la base de los
métodos ZAF, mientras que el segundo se utiliza en los métodos
f(rz). Si bien las
correcciones ZAF aún están disponibles en algunos sistemas/microsondas,
existe una tendencia general a reemplazarlas por correcciones
f(rz) más
avanzadas.
Aquí se considerarán de forma cualitativa
las correcciones ZAF. El factor de retrodispersión (Z) representa la
pérdida de intensidad de rayos X debido a los electrones
retrodispersados, el factor de absorción (A) corrige los rayos X
generados en la muestra pero que no pudieron escapar de ella y el
factor de fluorescencia (F) representa los rayos X generados no por
el impacto de electrones primarios, sino por la fluorescencia de rayos X
característicos primarios y de fotones de frenado. La corrección por
absorción suele ser la más significativa, especialmente en el análisis
de elementos ligeros, mientras que el factor de fluorescencia F suele
ser el menos significativo de los tres factores de corrección. Estos
factores tienen formulaciones matemáticas complejas que necesitan de
métodos de iteración para ser resueltas (Vea
Llovet et al., 2021 para las expresiones matemáticas y su
apéndice).
El factor de corrección "Z"
Los electrones incidentes pierden energía en interacciones inelásticas
con los electrones de la capa interna de la muestra dentro del volumen
analítico (o volumen de interacción entre los electrones del haz y la
muestra). Un material con un Z medio alto tiene una densidad más alta y
una configuración electrónica más compleja que un material con un Z
medio más bajo. Esto significa que, para el mismo voltaje de
aceleración, la profundidad a la que los electrones del haz pueden
penetrar en un material con Z medio alto es menor que la profundidad de
penetración de los materiales con un Z medio más bajo. El llamado "poder
de frenado" (energía perdida por los electrones del haz por unidad de
masa penetrada, o espectro continuo) no es constante, sino que disminuye
al aumentar Z. Esto tiene como consecuencia la producción de un mayor
número de electrones retrodispersados en materiales con Z medio alto
y, en consecuencia, un menor número de eventos de dispersión de rayos X.
Por otro lado, incluso con un menor número de interacciones de
dispersión de RX en los materiales con un Z más alto, las capas
electrónicas más complejas de los elementos con un Z más alto provocarán
una mayor cantidad y una familia más amplia de rayos X (por ejemplo,
valores significativos de Lα, Lβ, Mα, Mβ) en comparación con un elemento
con un Z más bajo. Si tanto Si como Fe están presentes en la misma
muestra con igual concentración, el número de rayos X producidos por uno
y otro sería diferente y, por lo tanto, el resultado de la primera
aproximación de Castaing debe corregirse. Esta corrección tiene dos
componentes: corrección por el "poder de frenado" y corrección por la "retrodispersión".
Si el Z medio de la muestra desconocida es mayor que el del estándar, se
debe aplicar una corrección a la baja en la composición.
Factor de corrección “A”
Algunos rayos X generados en el volumen analítico no escapan de la
muestra y son absorbidos por todos los elementos presentes en diferente
medida y en todas las direcciones de propagación. La absorción a lo
largo del camino hacia los detectores de rayos X y la longitud de dicho
camino requieren una corrección de la intensidad medida (es decir,
emitida) para obtener la intensidad inicial (es decir, generada) de los
rayos X. Esta corrección se denomina corrección de absorción (A).
Factor de corrección “F”
Los rayos X generados por un elemento dado, con una energía cercana o
superior a la energía crítica de excitación de otro elemento, al salir
del volumen analítico, pueden eliminar un electrón de una capa externa.
Dado que la vacante creada de esta manera siempre estará ocupada por un
electrón de una capa externa uniforme, se emitirá un fotón y, por lo
tanto, se producirá un rayo X. Este fenómeno se conoce como
fluorescencia secundaria. Por lo tanto, los rayos X de alta energía de
un elemento "i" fluorescen los rayos X del otro elemento "j" y acentúan
la intensidad de los rayos X del elemento "j" en comparación con la
producida únicamente por la excitación electrónica. La contribución
adicional de la fluorescencia por los rayos X característicos se
denomina F (corrección de fluorescencia).
Los parámetros Z, A y F se utilizan normalmente para multiplicar el
resultado obtenido por la "primera aproximación de Castaing". La llamada
"corrección ZAF" es Z*A*F, y se aplica tanto a los estándares como a la
muestra desconocida.
Si el material estándar tiene números ZAF idénticos a los números ZAF de
la muestra desconocida, el efecto de la corrección matricial se anulará
mutuamente (ZAFiunk = ZAFistd).
Por esta razón, en la práctica, se intenta elegir estándares del mismo
tipo de material que la muestra que analizamos como desconocida.

Lo que se hace es medir todos las razones k-ratios de todos los
elementos que componen la muestra (excepto los estimados por
estequiometría u otros métodos) y el sistema de ecuaciones resultante se
resuelve mediante un procedimiento iterativo. Esto es necesario entre
otras razones porque varias cantidades presentes en las ecuaciones que
describen los factores de corrección dependen de las concentraciones
desconocidas, que son las que se están calculando. En general, no
obstante, la convergencia se logra rápidamente mediante procedimientos
iterativos sencillos.
Por ejemplo, en un análisis de microsonda de un olivino (MgxFe₁-x)2SiO4
que contiene O, Mg, Si y Fe, es necesario corregir los efectos del
retardo y la dispersión de electrones (Z), la absorción de rayos X (A) y
la fluorescencia de rayos X (F) de todos los elementos sobre todos los
elementos. Se miden las intensidades de rayos X emitidos para las líneas
Kα de Mg, Si y Fe en una muestra de olivino y en los estándares,
calculándose las relaciones k para todos los elementos en relación con
los estándares utilizados para cada elemento. Por ejemplo, la
concentración de Mg en la muestra de olivino se obtiene multiplicando el
coeficiente k de Mg por la concentración de Mg en el estándar de Mg, y
todo se corrige multiplicando por los factores de corrección Z, A y F
para el Mg en la muestra y en el estándar. La conversión de k a C se
realiza mediante cálculo iterativo, inversión de matrices y otros tipos
cálculo. En cualquier caso, se puede determinar la concentración desde
el elemento traza hasta el elemento puro utilizando los algoritmos para
los factores ZAF.
4b.-
Análisis WDS y EDS mixtos y sin estándares.
Un detector de Espectrometría de Dispersión de Energía (EDS)
colocado dentro de la cámara de muestra (sobre la muestra) de una EPMA
puede identificar y discriminar los rayos X emitidos por su energía, así
como puede contar los rayos X de diferente energía. El análisis EDS
genera un espectro de intensidad de los rayos X característicos (número
de cuentas por segundo) en función de su energía característica,
generando picos en el espectro que se identifican y etiquetan
automáticamente con el elemento y el tipo de línea de rayos X (FeKα,
PbMβ, etc). Al igual que WDS, la altura del pico para el elemento "i" es
proporcional a la concentración del elemento "i" en la muestra, por lo
que al comparar la altura del pico con la de un estandard se puede calcular la concentración del elemento "i".
La calidad de los análisis WDS y EDS es parecida, aunque la precisión de
WDS es mayor.
Dado que el análisis EDS también
puede hacerse con estándares, al igual que en WDS,
comparando picos de la muestra desconocida con picos de los elemento medidos en el
estándar, es posible realizar un análisis cuantitativo
combinado donde algunos elementos se analizan en modo EDS y otros en
modo WDS, siempre que la instrumentación tenga un detector EDS integrado
en EPMA, o un espectrómetro de longitud de onda integrado en un SEM.
Así, aunque el análisis EPMA genérico basado en estándares aplica tanto
para el EPMA/SEM WDS como para el EPMA/SEM EDS, muchos análisis se
realizan sin que el usuario mida directamente los estándares,
especialmente con EDS. Esto es, se puede comparar la altura del pico
medido en la muestra con la de un "estándar virtual 100% puro". En el
análisis sin estándares, se utilizan intensidades de elementos puros
calculadas, derivadas de datos teóricos o empíricos contenidos en el
software, en lugar de valores medidos. Esta opción se ofrece en la
mayoría de los sistemas de dispersión de energía, pero su precisión es
difícil de evaluar, aunque ciertamente no es tan buena como cuando se
utilizan estándares reales.
La cuantificación sin estándares se basa en primeros principios, que
permiten simular la generación, emisión y detección de rayos X. Las
intensidades de los rayos X se calculan utilizando ecuaciones para una
composición de muestra y condiciones analíticas dadas. Esto requiere el
conocimiento no solo de magnitudes fundamentales, sino también de
parámetros instrumentales. En cualquier caso, se puede calcular o
extrapolar la intensidad de rayos X del elemento puro en unas
condiciones analíticas dadas y crear un estándar virtual. Este método es
muy susceptible a imprecisiones en los parámetros físicos y modelos,
considerándose un análisis semi-cuantitativo. De hecho, a menos
que la eficiencia del detector sea conocida y estable (es difícil de
predecir la eficiencia de los espectrómetros), los estándares virtuales
solo son aplicables de forma general a EDS. En los análisis sin
estándares genera valores de la concentración de los elementos (% en peso) siendo
la suma total de elementos lejana a 100%. Generalmente, el análisis se normaliza
al 100%, pero su calidad debe evaluarse mediante inspeccionando la
estequiometría resultante tras normalizar la composición a una fórmula
estructural.
4c.-
Comparación de EDS y WDS
Comparación práctica de EDS y WDS en términos de precisión de
microanálisis cuantitativo mediante
EDS y WDS en distintos
laboratorios de un vidrio metálico a base de níquel. Participaron 56
laboratorios, con 69 columnas de electrones y 75 espectrómetros. El
estudio se dividió en 56 microscopios electrónicos de barrido (SEM) y 13
sondas de electrones con confirguraciones variadas, 57 EDS (37
detectores SDD y 20 detectores de Si(Li)) y 18 sistemas WDS en 13 sondas
de electrones y 5 SEM. Las condiciones descritas aquí corresponden a
análisis bajo 15 o 20 kV.
El vidrio estaba compuesto de Ni (50,3 % en peso), Co (23,4 % en peso),
Cr (10,4 % en peso), Mo (6,7 % en peso), Fe (5,6 % en peso) y B (3,6 %
en peso).
El resultado clave de este estudio fue que, para cualquier laboratorio,
existe una probabilidad significativa de un mayor error analítico con
EDS que con WDS. La figura abajo muestra la dispersión de los resultados
para Ni y Mo. La mayoría de los resultados de Ni con WDS se sitúan
exactamente donde deberían (50-51 % en peso de Ni), en comparación con
la mayoría de los resultados de EDS que se sitúan en un rango más
amplio, entre el 51 % y el 53 % en peso. Hay tres veces más sistemas EDS
representados que sistemas WDS, por lo que cabría esperar una mayor
variación en los resultados de EDS, pero parece existir un problema de
precisión. Los resultados de WDS y EDS para Mo coincidieron o se
acercaron mucho a la composición objetivo, pero, una vez más, la
variabilidad en los resultados de EDS fue mayor de lo deseable.

Histogramas de los resultados de medición de Ni y Mo en el vidrio
mediante WDS y EDS en distintos laboratorios. A la izquierda, los
resultados de EDS muestran un error de 1-2 % en peso, sobrestimando el
contenido de Ni, en comparación con los resultados de WDS; además, los
valores determinados por EDS se distribuyen en un rango más amplio. A la
derecha, tanto EDS como WDS proporcionan valores de Mo correctos,
aunque, nuevamente, hay suficientes mediciones de EDS fuera de un rango
de error razonable (Jonnard et al., 2015).
Datos adicionales para la comparación EDS-WDS interlaboratorio se
ofrecen en la figura de abajo. Los datos para EDS se descompone en SDD y
Si(Li); para Ni, ambos dan generalmente resultados demasiado altos, con
más dispersión para
SDD en comparación con
Si(Li). Mo también muestra un
alto grado de dispersión. Por otro lado, los datos WDS de las sondas
electrónicas son mucho menos dispersos y más próximos a la composición
del material. La excepción son los detectores WDS montados en SEM: aquí
la dispersión se aproxima o supera a la de EDS. Las diferencias
instrumentales clave entre los SEM y las sondas electrónicas incluyen:
(i) posiblemente menos estabilidad del haz, (ii) potenciales errores en
el ángulo de despegue (deslizamientos del detector EDS hacia adentro o
hacia afuera, lo que potencialmente cambia el ángulo de despegue) y (iii)
sin un microscopio óptico en EDS para enfocar la superficie de la
muestra, la distancia de trabajo puede variar.

Resultados de 66 sistemas EDS y 18 sistemas WDS en distintos
laboratorios. La figura muestra datos de todos los elementos, así como
desgloses por tipo de EDS y la separación de los WDS en sondas
electrónicas en comparación con los WDS montados en microscopios
electrónicos de barrido (SEM). Los valores del eje x representan números
asignados arbitrariamente a los instrumentos.
Ruste J. Circuit d’intercomparaison 2012-2013. Group Natl Microsc Electron à Alayage Microanal 2013.
5a.- Análisis cerca de los límites de fase y el problema de la fluorescencia
secundaria
La región de la muestra desde la que se generan los rayos X primarios es
de varias micras cúbicas, pero la región donde se produce fluorescencia
secundaria puede ser de uno a dos órdenes de magnitud mayor. Esto
significa que incluso con un haz de electrones que incide a una
distancia considerable del límite con otra fase, puede haber rayos X
emitidos desde la fase adyacente que alcancen el detector. Esta
contribución, conocida como fluorescencia de límite o fluorescencia
secundaria (FS) cerca de los límites de fase, puede ser una fuente de
error significativa al analizar un elemento (particularmente
traza/menor) en una fase próxima a otra que contenga el elemento de
interés.
Por ejemplo, en la figura abajo se aprecia dos fases puras en contacto,
Cu y Co metálicos y que no han sido sometidos a procesos de inter-difusión
(o sea, Cu no contiene Co y Co no contiene Cu). Es claro que el
incremento de la concentración de uno en otro a medida que la sonda se
acerca al límite de fase es aparente. Más aún, el perfil aparente
registrado se parece a perfiles de difusión elemental, lo que puede
llevar a errores conceptuales importantes.
Este problema es agudo y difícil de resolver a la hora de analizar en
inclusiones pequeñas dentro de otras fases minerales. Tanto en este
caso como en otros relacionados con cercanía del límite de fase pueden
llevarse a cabo modelos matemáticos de sustracción de una fase en la
otra, si bien la calidad del análisis resultante calculado puede ser
deficiente.

Ejemplo de fluorescencia secundaria que imita la difusión elemental. En
este caso, los dos materiales puros se montan uno junto al otro, sin
difusión. Los códigos PENEPMA y FANAL ("este estudio") simulan con
precisión tanto la fluorescencia secundaria característica como la
continua. De
Llovet et al. (2012). Nótese que la distancia mínima para evitar la
contaminación del análisis es de unas decenas de micras.
5b.- Ejemplo de rutina analítica
Este ejemplo es para una Jeol Superprobre
| |
Element |
X-ray |
Crystal |
Spectrom |
Acc.v |
Peak Pos. (mm) |
(nm) |
BG_Low (mm) |
BG_Up (mm) |
Peak (s) |
BG (s) |
|
1 |
Si |
Ka |
TAP |
1 |
20 |
77.162 |
0.71254 |
7 |
5 |
10 |
5 |
|
2 |
Al |
Ka |
TAP |
1 |
20 |
90.475 |
0.83393 |
5 |
3.5 |
10 |
5 |
|
3 |
Mg |
Ka |
TAP |
1 |
20 |
107.453 |
0.989 |
5 |
3 |
10 |
5 |
|
4 |
Cr |
Ka |
PETJ |
2 |
20 |
73.099 |
0.22897 |
2.5 |
2.5 |
10 |
5 |
|
5 |
Ti |
Ka |
PETJ |
2 |
20 |
87.805 |
0.27485 |
2.6 |
5 |
10 |
5 |
|
6 |
Ca |
Ka |
PETJ |
2 |
20 |
107.336 |
0.33584 |
4 |
6 |
10 |
5 |
|
7 |
V |
Ka |
LIFL |
3 |
20 |
173.969 |
0.25036 |
3 |
3 |
10 |
5 |
|
8 |
Co |
Ka |
LIFL |
3 |
20 |
124.198 |
0.1789 |
4.4 |
3 |
10 |
5 |
|
9 |
Zn |
Ka |
LIFL |
3 |
20 |
99.495 |
0.14352 |
5 |
2.5 |
10 |
5 |
|
10 |
Mn |
Ka |
LIFH |
4 |
20 |
146.131 |
0.21018 |
4 |
3 |
10 |
5 |
|
11 |
Fe |
Ka |
LIFH |
4 |
20 |
134.603 |
0.1936 |
4 |
3 |
10 |
5 |
|
12 |
Ni |
Ka |
LIFH |
4 |
20 |
115.201 |
0.16579 |
5 |
3 |
10 |
5 |
| |
|
|
|
|
|
|
|
|
|
|
|
|
Measurement |
Order |
of |
WDS |
|
|
|
|
|
|
|
|
Order |
Channel 1 |
2 |
3 |
4 |
|
|
|
|
|
|
|
|
1 |
Si_CH1_T |
Cr_CH2_P |
V_CH3_LI |
Mn_CH4_L |
|
|
|
|
|
|
|
|
2 |
Al_CH1_T |
Ti_CH2_P |
Co_CH3_L |
Fe_CH4_L |
|
|
|
|
|
|
|
|
3 |
Mg_CH1_T |
Ca_CH2_P |
Zn_CH3_L |
Ni_CH4_L |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Calc. |
Elements : |
O |
(Anion) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Standard |
Data |
|
|
|
|
|
|
|
|
|
|
| |
Element |
Standard |
|
|
Mass(%) |
|
|
|
|
|
|
|
1 |
SiO2 |
Diopside |
CaMgSi2O6 |
|
55.1912 |
|
|
|
|
|
|
|
2 |
Al2O3 |
Kyanite |
Al2SiO5 |
|
62.6952 |
|
|
|
|
|
|
|
3 |
MgO |
Chromite |
(Mg,Fe++)(Al,Cr)2O4 |
17.2615 |
|
|
|
|
|
|
|
4 |
Cr2O3 |
Chromite |
(Mg,Fe++)(Al,Cr)2O4 |
45.6438 |
|
|
|
|
|
|
|
5 |
TiO2 |
Rutile |
TiO2 |
|
100.0001 |
|
|
|
|
|
|
|
6 |
CaO |
Wollastonite |
CaSiO3 |
|
47.7408 |
|
|
|
|
|
|
|
7 |
V2O3 |
V |
V |
|
147.1143 |
|
|
|
|
|
|
|
8 |
CoO |
Co |
Co |
|
127.1509 |
|
|
|
|
|
|
|
9 |
ZnO |
Sphalerite |
(Zn,Fe)S |
|
83.5234 |
|
|
|
|
|
|
|
10 |
MnO |
Rhodonite |
(Mn++,Fe++,Mg,Ca)SiO3 |
43.4885 |
|
|
|
|
|
|
|
11 |
FeO |
Fe2O3 |
Fe2O3 |
|
89.9765 |
|
|
|
|
|
|
|
12 |
NiO |
NiO |
NiO |
|
99.9951 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Standard Intensity of WDS |
|
|
|
|
|
|
|
|
|
| |
Element |
Curr.(A) |
Net(cps) |
Bg-(cps) |
Bg+(cps) |
S.D.(%) |
Date |
|
|
|
|
|
1 |
Si |
9.73E-09 |
7819.3 |
33.3 |
23.7 |
0.48 |
30/01/2024 |
9:52:51 |
|
|
|
|
2 |
Al |
9.62E-09 |
9521.7 |
31.1 |
20.2 |
0.44 |
30/01/2024 |
9:23:37 |
|
|
|
|
3 |
Mg |
1.45E-08 |
1874.8 |
19.8 |
17.2 |
0.99 |
30/01/2024 |
9:59:10 |
|
|
|
|
4 |
Cr |
1.45E-08 |
4592.9 |
56.6 |
46 |
0.63 |
30/01/2024 |
9:59:10 |
|
|
|
|
5 |
Ti |
9.75E-09 |
7017.3 |
24.8 |
17.5 |
0.51 |
30/01/2024 |
9:49:36 |
|
|
|
|
6 |
Ca |
9.73E-09 |
3901.8 |
11.4 |
8.6 |
0.68 |
30/01/2024 |
9:55:57 |
|
|
|
|
7 |
V |
9.67E-09 |
5963.6 |
9.4 |
9.4 |
0.55 |
30/01/2024 |
10:12:17 |
|
|
|
|
8 |
Co |
9.70E-09 |
12209.6 |
35.7 |
32.6 |
0.39 |
30/01/2024 |
10:05:45 |
|
|
|
|
9 |
Zn |
9.70E-09 |
8109.2 |
55.3 |
51.8 |
0.47 |
30/01/2024 |
10:08:58 |
|
|
|
|
10 |
Mn |
1.46E-08 |
6968.2 |
34.4 |
33.6 |
0.51 |
30/01/2024 |
9:46:04 |
|
|
|
|
11 |
Fe |
9.68E-09 |
11653.6 |
40.9 |
43.1 |
0.39 |
30/01/2024 |
9:42:45 |
|
|
|
|
12 |
Ni |
1.45E-08 |
21679.6 |
110.7 |
110.9 |
0.29 |
30/01/2024 |
10:02:32 |
|
|
|
5c.-
Criterios de exactitud (calidad)
Para evaluar la exactitud de los resultados de los análisis basados en
estándares, tanto en res, tanto en WDS como EDS (utilizando explícitamente un material
de referencia local para adquirir los recuentos de rayos X y generar la
relación k requerida), se utiliza una prueba sencilla: ¿el porcentaje de
masa (peso) analítico total se encuentra en un rango cercano al 100 % en
peso (sin normalizar a 100)? Idealmente, un total analítico "perfecto"
debería estar entre el 99 y el 100,5 % en peso (un valor más bajo, ya
que podría haber elementos sin medir, como el
hidrógeno en minerales hidratados, con OH- o H2O
molecular, o elementos minoritarios/trazas), aunque en la práctica se considera aceptable un margen más
amplio.
Existe un segundo criterio de control de calidad, aplicable a algunos
materiales en los que se sabe que los elementos existen en una relación estequiométrica bien definida. Sin embargo, esto puede ser complicado
cuando existe un rango de solución sólida.
Existe un tercer criterio de equilibrio de carga para un material con
enlace covalente.
Hay que indicar que todos los criterios anteriores pueden superarse en
el caso de problemas relacionados con la cercanía de límites de fase.
Esto es, dos fases en contacto analizadas cerca del mismo darán señales
aparente de tener disuelta la fase adyacente en cierta cantidad
(pudiendo ser o no solución sólida), la suma elemental será cercana al
100% (si las dos fases no contienen hidrógeno y si han analizado todos
los elementos significativos) y las cargas estarán balanceadas (si se
analizan todos los aniones y cationes significativos en materiales
iónicos y covalentes).
Puede haber un problema de control de calidad cuando los usuarios de
EPMA "solo" miran los valores de porcentaje atómico normalizado.
El problema es que, si el total analítico está lejos del 100 % en peso,
eso es una señal de alerta inmediata de que hay problemas con el
análisis, por ejemplo, un elemento no analizado en el desconocido (total
bajo), un estándar dañado (total alto) o fluorescencia secundaria de
fases adyacentes (total alto). Esta señal de alerta indica que no se
puede asumir que los valores atómicos normalizados sean correctos. Por
esa razón, el total de porcentaje en peso no normalizado
siempre debe consultarse antes de aceptar los valores atómicos
normalizados, y los informes siempre deben incluir los totales
analíticos (% en peso) sin normalizar.

Control de calidad.
5d.-
Aplicaciones inadecuadas
En ocasiones, los usuarios de WDS o EDS intentan utilizar el instrumento
y el proceso analítico de forma inadecuada. A continuación, se presentan
tres ejemplos basados en experiencias reales de
laboratorio:
-
“Solo me interesa un elemento de esta muestra, así
que, por favor, no mida ningún otro”: El material obviamente
contiene más de un elemento, por lo que la intensidad de rayos X
medida para el elemento “objetivo” se ve modificada por la matriz.
Por lo tanto, TODOS los elementos presentes en la muestra DEBEN
medirse (o al menos tenerse en cuenta) para determinar la
concentración correcta del elemento objetivo.ento objetivo.
-
Una variante de esto es la creencia de que se
puede medir mediante WDS o EDS la composición de una película
delgada sobre un sustrato “ignorando los elementos del sustrato y
renormalizando los elementos de la película”. Nuevamente, esto puede
generar errores, ya que el haz de electrones penetra la película y,
por lo tanto, la "ve" en una fracción de la profundidad que recorren
los electrones. Por lo tanto, esos rayos X no se generan de forma
similar a como se generan dentro del material de referencia. Por lo
tanto, las relaciones k de los elementos de la película son
"incorrectas" incluso antes de que entren en la corrección de la
matriz. A esto se suma la posibilidad de que los rayos X en el
sustrato provoquen fluorescencia secundaria en la película,
generando más errores.
Existe una forma correcta de determinar la composición de películas
delgadas mediante EDS y WDS.
-
Análisis de haz desenfocado: En ocasiones, se
utiliza EPMA para intentar lograr un análisis de un área
relativamente grande. El volumen de interacción de los haces de
electrones incidentes de 15-20 kV es del orden de 1-2 µm,
dependiendo de la composición y la densidad del objetivo. Sin
embargo, a veces se solicita a un laboratorio la determinación de la
composición química de una pequeña región heterogénea/multifásica
dentro de una muestra. Dado que el haz de electrones en modo puntual
puede desenfocarse o configurarse en modo de escaneo rectangular, se
puede creer que estos modos de desenfocado/escaneo pueden recolectar
rayos X de un área extensa (de hasta 50–100 µm de ancho), lo que
produce una composición química promedio correcta. Sin embargo, esta
aplicación de EPMA es propensa a errores, ya que viola un principio
clave del procedimiento de corrección (rayos X → relación k →
corrección matricial → resultado para cada volumen analítico
discreto), que es que el volumen de interacción es homogéneo, de
modo que se aplica una corrección única. Si el haz genera rayos X en
más de una composición química, la corrección matricial promediará
los factores de corrección, generando errores. Dado que en muchas
situaciones la corrección por absorción puede ser la corrección más
importante, el sobreescaneo en muchos casos genera totales
sintomáticamente altos. El procedimiento adecuado es adquirir
mapas de rayos X, cuantitaticarlos, y luego
procesarlos para generar composiciones globales correctas áreas
pequeñas o grandes.
5.e.- Directrices para la presentación de resultados de EPMA/SEM
Las publicaciones de investigación y reportes técnicos deben
proporcionar una descripción de las técnicas utilizadas y los
procedimientos analíticos seguidos para la obtención de los datos
reportados (mediante EDS o WDS), incluyendo, a ser posible:
-
Instrumento utilizado (fabricante y modelo),
voltaje de aceleración, corriente del haz (si se mide) y si el haz
está enfocado o desenfocado (y su tamaño).
-
Para EDS: marca y modelo del detector de energía;
tipo de ventana, ±(constante de tiempo, tiempo muerto, ángulo de
despegue, software).
-
Para WDS: cristales específicos utilizados para la
difracción de RX, especialmente para elementos ligeros, ±(valor de
espaciado 2d; analizador de altura de pulso (PHA); ángulo de
despegue; software).
-
Preparación de la muestra: nivel de pulido
±(conductividad eléctrica), material de recubrimiento ±(espesor).
-
Estándares para cada elemento medido.
-
Si se determinaron elementos por diferencia,
estequiometría u otro método (e.g., H, O,..).
-
Tiempo de contaje (línea de rayos X y fondos),
error analítico y límites de detección de trazas.
-
Qué corrección matricial se utilizó (es decir, qué
ZAF específico o qué modelo se utilizó)
-
±(Líneas de rayos X medidas/utilizadas, posiciones
(energías) donde se midieron los valores de fondo (si se midieron),
modelo utilizado para describir el fondo (lineal, exponencial,
pendiente, multipunto, MAN).
-
±(Si se utilizaron procedimientos específicos:
anticontaminación; estrategia de conteo dependiente del tiempo;
correcciones por interferencias).
-
±(Si se trata de elementos ligeros, fuente de los
MAC utilizados).
-
Directrices utilizadas para el control de calidad
de los datos (p. ej., totales analíticos no normalizados, totales
analíticos excluidos por debajo o por encima de un umbral de % en
peso determinado, otros criterios).
-
Los totales analíticos no normalizados del % en
peso deben proporcionarse como comprobación analítica, además de
fórmulas estructurales.
-
Detalles experimentales de los mapas de RX
(elementales). Condiciones, cuantificación en su caso, etc.
5f.-
Ejemplos
Ejemplo tomado de
García-Casco et al. (2025) A general conceptual petrological model for the subsolidus transformation of chromitite with application and implications. Journal of Petrology, 66(4),
egaf029 > DATA in Interdisciplinary Earth Data Alliance (IEDA)
Back-scattered electron (BSE) images, quantitative electronprobe
microanalyses and X-ray (XR) elemental maps were obtained with a JEOL
JXA-8230 at the Centres Científics i Tecnològics, Barcelona University (CCiTUB),
operated in wavelength-dispersive spectroscopy (WDS) mode. Operating
conditions for point analyses were 20 kV acceleration potential, 15 nA
beam current, focused beam and 20 seconds/element (see Pujol-Solà et al.,
2021 for further details).
Spinel analyses were normalized to 4
oxygens and 3 cations, while the analyses of chlorite were normalized to
20 oxygens, 16(OH) and 20 cations (Supplementary
Table S1 and EarthChem Library link). Fe3+ content
was estimated by stoichiometry. Atoms per formula unit is abbreviated
apfu.
X-ray maps were collected using the EPMA operated in WDS mode by
stage scanning, with an accelerating voltage of 20 kV, beam current of
300 nA, dwell time of 20 ms per pixel, and pixel sizes (point spacing)
of 0.38 to 2.5 μm. Experiments have demonstrated that short counting
time (milliseconds rather than seconds) precludes beam damage at high
beam current (García-Casco,
2007).
The raw counts were transformed into oxide wt % with the ZAF correction.
All maps were converted into molar oxide and atomic proportions and
manipulated with DWImager software (see García-Casco,
2007;
Torres-Roldán & Garcia-Casco, N.D.). This software was used to extract
the bulk-compositions of the scanned areas and micro-domains.
Reactions
and tetrahedral and triangular projections have been obtained using
algebraic methods (matrix analysis; e.g. Spear et al.,
1982)
with CSpace (Torres-Roldan et al.,
2000;
see also Garcia-Casco et al.,
2020)
and Excel softwares. In the end-member system MgO–Al2O3–SiO2–H2O
(MASH) (system components = 4), stoichiometric coefficients of reactions
among 5 selected phases (e.g. forsterite, spinel, chlorite, brucite and
H2O
fluid, eq. 6 below)
are straightforwardly found by matrix analysis. Bulk rock compositions
in the 6-component system FeO–MgO–Al2O3–Cr2O3–SiO2–H2O
(FMACrSH) were algebraically mapped in new 6-dimensional coordinate
systems defined by Mg–Al-bearing mineral end-members, exchange vectors
Cr2O3(Al2O3)−1 and
FeO(MgO)−1 (hereafter
abbreviated FeMg−1 and
CrAl−1,
respectively) and H2O fluid, such as in eq. 11.
Whole-rock compositions developed upon metasomatic MgO-loss from, or SiO2-addition
in, model chromitite (0.8:0.2 spinel/olivine ratio, molar units, WR0
below) were calculated in the FMACrSH system iteratively until an
assemblage made of spinel and chlorite is reached (e.g. eq. 18 below).
Most calculations were performed in molar units, but some are offered in
oxy-equivalent units (e.g. eq. 19 below)
to approximate the volume proportions of the solid phases (Brady
& Stout, 1980; Thompson,
1982; Garcia-Casco et al.,
2020).
These calculations were combined with calculations involving Fe–Mg and
Al–Cr exchanges between olivine and spinel and chlorite and spinel using
varied distribution coefficients (KDs),
as justified below.
Triangular and tetrahedral phase diagrams for the 6-component
FMACrSH system involve projection from phases and exchange vectors, as
indicated in the corresponding figures. Thermodynamic calculations have
been performed using GeoPS v3.5.4 (Xiang
& Connolly, 2022)
and the thermodynamic dataset HP633 (Holland
& Powell, 2011)
and associated solutions models for olivine, orthopyroxene, spinel and
garnet (Holland et al.,
2018),
plus chlorite, chloritoid, staurolite, cordierite (White et al.,
2014)
and talc (ideal Fe–Mg solution) and H2O-fluid
(Holland
& Powell, 1998).
Mineral abbreviations are after Warr
(2021).
Ejemplo tomado de Domínguez-Carretero
et al 2024. The Geology, Geochemistry, and Mineralogy of the Moa Bay Ni Laterite Mining District, Cuba. Economic Geology 119 (7), 685-1706.
Electron
microprobe analyses on oxyhydroxides (goethite, maghemite, Mn
oxyhydroxides) and silicates (serpentine and garnierite) were conducted
at CCiT-UB with a JEOL JXA-8230 equipment with four wavelength-dispersive
spectrometers (WDS) and one EDS system (App. 5). The analytical
conditions were 15- to 20-kV accelerating voltage, 10- to 20-nA beam
current, 1- to 2-μm beam diameter, and 10- to 20-s counting time (peak
and background) per element. Measurements and calibrations were
performed using the following natural and synthetic standards: hematite
(Fe), rutile (Ti), periclase (Mg), rhodonite (Mn), Al2O3 (Al),
Cr2O3 (Cr), diopside (Si), NiO (Ni), wollastonite
(Ca), orthoclase (K), metallic cobalt (Co), and sphalerite (Zn).
A special
EMP procedure was designed for measuring Sc in goethite, maghemite, Mn
oxyhydroxides, and hematite from the Punta Gorda and Yamanigüey deposits.
Scandium was measured first with four WDS instruments and one EDS
instrument for 12 min of counting time (peak and background), followed
by the analysis of Si, Al, Cr, Mg, Ti, Co, Ni, Fe, and Mn with counting
times of 10 to 20 s (peak and background), for a 20-kV accelerating
voltage and 15-nA beam current with a probe size of 5 μm. The standards
used were the same as for the procedure used for major elements in
oxyhydroxides and silicates, plus metallic scandium to calibrate Sc. The
detection limit for Sc for these analyses is 15 ppm. Maghemite and
hematite Sc measurements were all below the limit of detection.
The
structural formulae of serpentine group minerals were calculated on the
basis of 7 oxygens and that of garnierites on the basis of 7 or 11
oxygens, depending on the predominance of serpentine or kerolite-pimelite
(talc) in the mixture. The talc fraction or Xtalc in the
mixture was calculated using the method of Villanova-de-Benavent et al.
(2022).
Iron in primary hydrothermal serpentine (“serpentine I”) was calculated
as Fe2+, and in secondary weathering serpentine (“serpentine
II”) and in garnierites as Fe3+ (Roqué-Rosell
et al., 2017; Villanova-de-Benavent
et al., 2017).
Ejemplo tomado de
Pujol-Solà et.
al. (2021). The chromitites of the Neoproterozoic Bou Azzer ophiolite (Central Anti-Atlas, Morocco) revisited. Ore Geology Reviews 134,
104166.
We studied representative samples from three
chromitite mining sites in the Bou Azzer ophiolite: Inguijem, Filon 60
and Ait Ahmane (Fig.
1, Fig.
2), including massive chromitites from individual pods (n = 4), semi-massive
chromitites (n = 2), banded chromitites (n = 1) and the host dunites.
Polished thin sections were studied by optical and electron
microscopy using a Quanta 200 FEI XTE 325/D8395 scanning electron
microscope (SEM) equipped with an INCA Energy 250 EDS microanalysis
system and a JEOL JSM-7100 field-emission SEM at the Centres Científics
i Tecnològics de la Universitat de Barcelona (CCiTUB). Operating
conditions were 20 kV accelerating voltage. Micro-Raman spectra were
obtained with a HORIBA JobinYvon LabRam HR 800 dispersive spectrometer
equipped with an Olympus BXFM optical
microscope at the CCiTUB. Non polarized Raman
spectra were obtained in confocal geometry by applying a 532 nm
laser, using a 100x objective (beam size around 2 µm), with 3–5
measurement repetitions for 10–15 s each. The Si band at ~520 cm−1 was
used for calibration. The obtained spectra were processed using the
LabSpec® software (JobinYvon; Villeneuve-d’Ascq, France).
Quantitative electron microprobe analyses (EMPA) on
chromite and platinum group minerals (PGM) were also conducted at the
CCiTUB using a JEOL JXA-8230 operating in wavelength-dispersive
spectroscopy (WDS) mode. Analytical conditions are described in detail
in Appendix 1. X-ray (XR) images of chromite (512 × 512 pixels,
0.52 × 0.52 µm pixel size and 266.24 × 266.24 µm total area) and
platinum group minerals (three images of 300 × 300 pixels: 0.22 µm pixel
size and 66 × 66 µm total area, 0.08 µm pixel size and 24 × 24 µm total
area and 0.1 µm pixel size and 30 × 30 µm total area) were obtained with
the same a JEOL JXA-8230 electron microprobe operated at 20 kV and 20nA
in spot mode with focused electron
beam and counting time of 20 to 60 ms/pixel. The images were treated
with DWImager software (Torres-Roldán & Garcia Casco, unpublished; see Garcia-Casco,
2007). Minor and trace elements in chromite were analyzed using a
Resolution M-50 Excimer laser
coupled to a ThermoICap Qc inductively coupled mass spectrometer at the
Laboratorio de Estudios Isotópicos (LEI) from the Centro de Geociencias,
UNAM (Mexico). The analytical conditions and procedure are described in
Appendix 1.
Appendix 1.
Electron microprobe analyses (EMPA)
Quantitative analyses of chromite and platinum group minerals (PGM) were
performed using a five-channel JEOL JXA-8230 electron microprobe at the
Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB).
The analytical conditions were 15 - 20 kV accelerating voltage, 10 - 20
nA beam current, 1 - 2 µm beam diameter, and 10 - 20 s counting time per
element, using WDS detectors and XPP matrix correction (Pouchou and Pichoir, 1990). The results for chromite were
stoichiometrically recalculated to distinguish FeO and Fe2O3
according to the procedure described by
Carmichael (1967). The measurements and the calibrations for
chromite and PGM analyses were performed using natural and synthetic
standards and are shown in Tables A and B.
|
Table A. Standards
used for chromite measurements on EMPA. |
|
Element |
Standard |
X-ray |
Crystal |
Channel |
|
|
Si |
Diopside |
Ka |
TAP |
1 |
|
|
Al |
Kyanite |
Ka |
TAP |
1 |
|
|
Cr |
Cr2O3 |
Ka |
PETJ |
2 |
|
|
Ti |
Rutile |
Ka |
PETJ |
2 |
|
|
Mg |
Periclase |
Ka |
TAPH |
3 |
|
|
Co |
Co |
Ka |
LIFH |
4 |
|
|
Fe |
Fe2O3 |
Ka |
LIFH |
4 |
|
|
V |
V |
Ka |
LIFH |
4 |
|
|
Ni |
NiO |
Ka |
LIFL |
5 |
|
|
Zn |
Sphalerite |
Ka |
LIFL |
5 |
|
|
Mn |
Rhodonite |
Ka |
LIFL |
5 |
|
|
Table B. Standards
used for PGM measurements on EMPA. |
|
Element |
Standard |
X-ray |
Crystal |
Channel |
|
|
Ru |
Ru |
La |
PETJ |
1 |
|
|
Rh |
Rh |
La |
PETJ |
1 |
|
|
Pd |
Pd |
Lb |
PETJ |
1 |
|
|
S |
Chalcopyrite |
Ka |
PETJ |
2 |
|
|
As |
GaAs |
Lb |
TAPH |
3 |
|
|
Cr |
Cr2O3 |
Ka |
LIFH |
4 |
|
|
Fe |
Chalcopyrite |
Ka |
LIFH |
4 |
|
|
Co |
Co |
Ka |
LIFH |
4 |
|
|
Ni |
Ni |
Ka |
LIFH |
4 |
|
|
Cu |
Chalcopyrite |
Ka |
LIFL |
5 |
|
|
Pt |
Pt |
La |
LIFL |
5 |
|
|
Ir |
Ir |
La |
LIFL |
5 |
|
|
Os |
Os |
La |
LIFL |
5 |
|
|
